

Нужен ли производителю медицинских изделий Сертификат CE для продажи в Европейском Союзе или это должен быть Сертификат CE? Данная статья поможет разобраться.
Читать дальше>>

Программа MDSAP позволяет производителям, пройдя единственный аудит, получить сертификат соответствия СМК для пяти стран.
Читать дальше>>

510(k) - это уведомительная регуляторная процедура, направленная на получение одобрения FDA на выведение медицинского изделия на рынок США.
Читать дальше>>
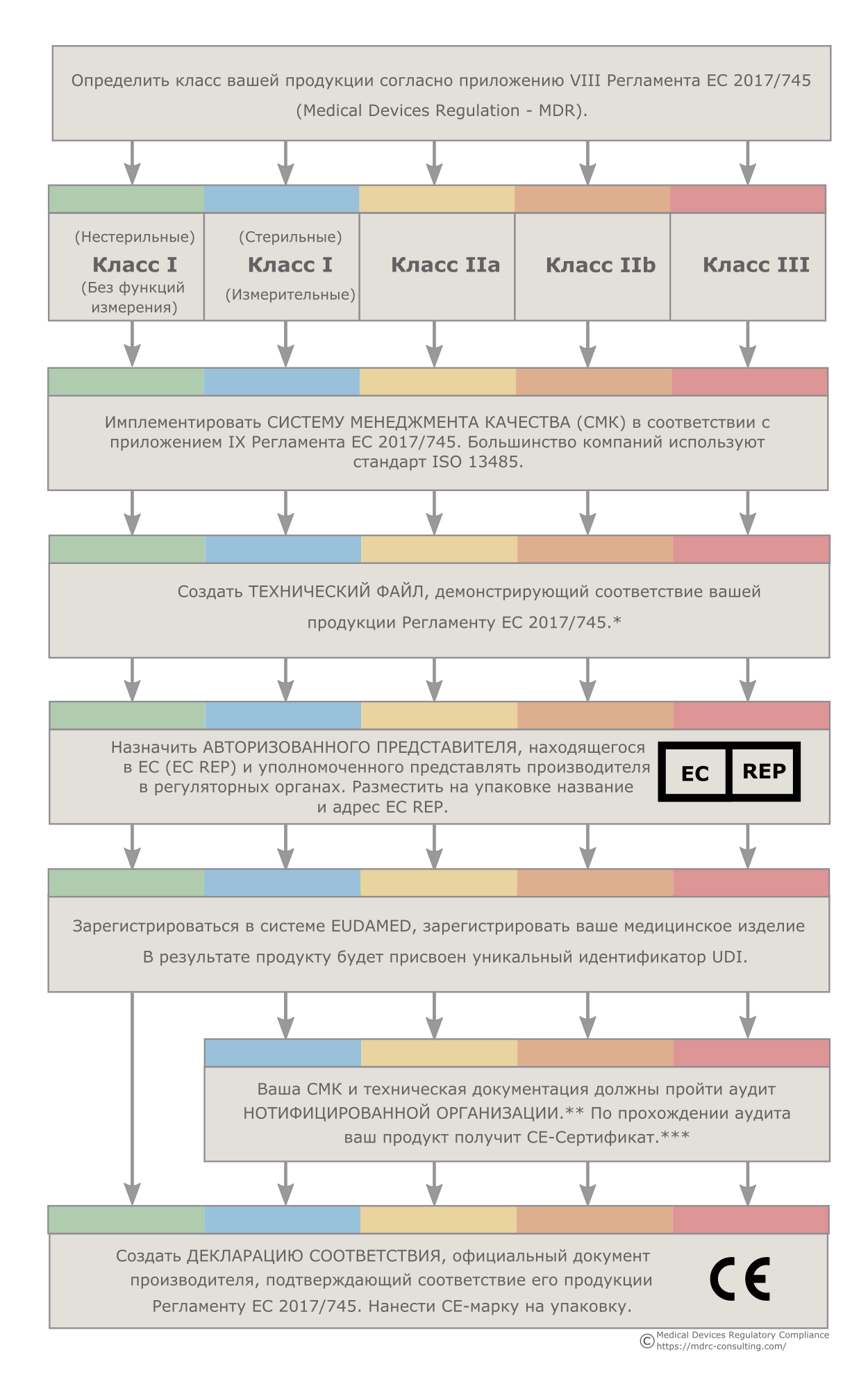
Чтобы зарегистрировать медицинское изделие в ЕС необходимо выполнить ряд требований европейского законодательства. В статье приводится пошаговая инструкция...
Читать дальше>>

Базовый UDI-DI - это одно из новых требований европейского законодательства в области медицинских изделий, связанное с вступлением в силу европейских регламентов...
Читать дальше>>

СЕ-маркировка для медицинских изделий равноценна разрешению на продажу медицинского изделия на...
Читать дальше>>

Согласно требованиям регламента ЕС MDR 2017/745, каждый производитель медицинских изделий должен иметь в своем распоряжении PRRC - лицо, отвечающее...
Читать дальше>>

Определение класса медицинского изделия - важнейший шаг на пути к получению одобрения...
Читать дальше>>

MDSAP - Medical Device Single Audit Program. Единая унифицированная программа аудита системы качества - инфографика в PDF-формате.
Читать дальше>>
Все производители изделий для in vitro диагностики, продающие свою продукцию в Европе, должны обеспечить соответствие европейскому регламенту IVDR 2017/746.
Читать дальше>>

Основные требования к регистрационной документации для медицинских изделий...
Читать дальше>>

В Эквадоре процесс регистрации медицинских изделий определяется несколькими основными нормативно-правовыми...
Читать дальше>>

Регистрация медицинских изделий в Коста-Рике осуществляется в два этапа. В состав регистрационного досье входят следующие документы...
Читать дальше>>

Регистрация в EUDAMED - обязательное условие, которое должны выполнять экономические операторы, обеспечивающие обращение медицинских изделий в Европейском Союзе.
Читать дальше>>
Медицинские маски могут использоваться как с профессиональными (медицинским персоналом), так и с непрофессиональными (например, жителями городов) целями. В связи с этим у производителя...
Читать дальше>>

Для любой компании, работающей с медицинскими изделиями ISO 13485 является...
Читать дальше>>
В ЕС, США и странах Латинской Америки средства для дезинфекции могут рассматриваться как изделия медицинского назначения, как биоциды или как лекарственные средства.
Читать дальше>>

Мировой рынок медицинских интернет технологий по оценкам экспертов к 2022 году достигнет 158,1 млрд долларов. За следующие два года аналитики предсказывают увеличение его размеров в 2-4 раза.
Читать дальше>>

Программа аудита системы качества MDSAP позволяет производителям изделий медицинского назначения по результатам единственного аудита получать сертификат, признаваемый регуляторными органами целого ряда стран.
Читать дальше>>

Для регистрации изделия медицинского назначения класса III или IV в Бразилии в составе регистрационного досье необходимо подать сертификат соответствия BGMP...
Читать дальше>>

Требования Руководства MDCG 2019-16 обязательны для производителей электронного медицинского оборудования. Руководство поможет производителям достичь регуляторного соответствия в ЕС.
Читать дальше>>

Новые нормативы Евросоюза требуют от производителей медицинской продукции предоставления клинических данных. Клинические исследования изделий медицинского назначения в ЕС...
Читать дальше>>

Критерии информационной безопасности медицинского оборудования FDA предназначены для производителей медицнского ПО и электронного медицинского оборудования, продающих в США...
Читать дальше>>

Одноразовые медицинские перчатки могут являться изделием медицинского назначения, средством индивидуальной защиты или и тем и тем одновременно.
Читать дальше>>

Регламент EC 2017/745 (MDR) влияет на регулирование рынка изделий медицинского назначения стран за пределами ЕС.
Читать дальше>>

Если вы хотите продавать медицинские изделия в ЕС, вам придется выбрать Нотифицированную Организацию...
Читать дальше>>

Соответствие 21 CFR Part 11 обязательное требование FDA к производителям медицинского оборудования, использующим электронные записи и подписи.
Читать дальше>>
GSPR — требования, которые обязан выполнять каждый производитель медицинских изделий, продающий свою продукцию в ЕС, независимо от вида и класса продукции.
Читать дальше>>


Нужен ли производителю медицинских изделий Сертификат CE для продажи в Европейском Союзе или это должен быть Сертификат CE? Данная статья поможет разобраться.
Читать дальше>>
Программа MDSAP позволяет производителям, пройдя единственный аудит, получить сертификат соответствия СМК для пяти стран.
Читать дальше>>
510(k) - это уведомительная регуляторная процедура, направленная на получение одобрения FDA на выведение медицинского изделия на рынок США.
Читать дальше>>
Чтобы зарегистрировать медицинское изделие в ЕС необходимо выполнить ряд требований европейского законодательства. В статье приводится пошаговая инструкция...
Читать дальше>>
Базовый UDI-DI - это одно из новых требований европейского законодательства в области медицинских изделий, связанное с вступлением в силу европейских регламентов...
Читать дальше>>
СЕ-маркировка для медицинских изделий равноценна разрешению на продажу медицинского изделия на...
Читать дальше>>
Согласно требованиям регламента ЕС MDR 2017/745, каждый производитель медицинских изделий должен иметь в своем распоряжении PRRC - лицо, отвечающее...
Читать дальше>>
Определение класса медицинского изделия - важнейший шаг на пути к получению одобрения...
Читать дальше>>

MDSAP - Medical Device Single Audit Program. Единая унифицированная программа аудита системы качества - инфографика в PDF-формате.
Читать дальше>>
Все производители изделий для in vitro диагностики, продающие свою продукцию в Европе, должны обеспечить соответствие европейскому регламенту IVDR 2017/746.
Читать дальше>>
Основные требования к регистрационной документации для медицинских изделий...
Читать дальше>>
В Эквадоре процесс регистрации медицинских изделий определяется несколькими основными нормативно-правовыми...
Читать дальше>>
Регистрация медицинских изделий в Коста-Рике осуществляется в два этапа. В состав регистрационного досье входят следующие документы...
Читать дальше>>
Регистрация в EUDAMED - обязательное условие, которое должны выполнять экономические операторы, обеспечивающие обращение медицинских изделий в Европейском Союзе.
Читать дальше>>
Медицинские маски могут использоваться как с профессиональными (медицинским персоналом), так и с непрофессиональными (например, жителями городов) целями. В связи с этим у производителя...
Читать дальше>>
Для любой компании, работающей с медицинскими изделиями ISO 13485 является...
Читать дальше>>

В ЕС, США и странах Латинской Америки средства для дезинфекции могут рассматриваться как изделия медицинского назначения, как биоциды или как лекарственные средства.
Читать дальше>>
Мировой рынок медицинских интернет технологий по оценкам экспертов к 2022 году достигнет 158,1 млрд долларов. За следующие два года аналитики предсказывают увеличение его размеров в 2-4 раза.
Читать дальше>>
Программа аудита системы качества MDSAP позволяет производителям изделий медицинского назначения по результатам единственного аудита получать сертификат, признаваемый регуляторными органами целого ряда стран.
Читать дальше>>
Для регистрации изделия медицинского назначения класса III или IV в Бразилии в составе регистрационного досье необходимо подать сертификат соответствия BGMP...
Читать дальше>>
Требования Руководства MDCG 2019-16 обязательны для производителей электронного медицинского оборудования. Руководство поможет производителям достичь регуляторного соответствия в ЕС.
Читать дальше>>
Новые нормативы Евросоюза требуют от производителей медицинской продукции предоставления клинических данных. Клинические исследования изделий медицинского назначения в ЕС...
Читать дальше>>
Критерии информационной безопасности медицинского оборудования FDA предназначены для производителей медицнского ПО и электронного медицинского оборудования, продающих в США...
Читать дальше>>
Одноразовые медицинские перчатки могут являться изделием медицинского назначения, средством индивидуальной защиты или и тем и тем одновременно.
Читать дальше>>
Соответствие 21 CFR Part 11 обязательное требование FDA к производителям медицинского оборудования, использующим электронные записи и подписи.
Читать дальше>>
GSPR — требования, которые обязан выполнять каждый производитель медицинских изделий, продающий свою продукцию в Европейском Союзе, независимо от вида и класса продукции.
Читать дальше>>
Если вы хотите продавать медицинские изделия в ЕС, вам придется выбрать Нотифицированную Организацию...
Читать дальше>>
Регламент EC 2017/745 (MDR) влияет на регулирование рынка изделий медицинского назначения стран за пределами ЕС.
Читать дальше>>
Мы будем рады обсудить ваш новый проект!

