Рынок медицинских изделий Европейского Союза строго регулируется. Если вы хотите продавать свою продукцию медицинского назначения в Европе, вам необходимо выполнить ряд требований, которые европейские регуляторы предъявляют к продуктам такого рода.
Иногда этот процесс называют регистрацией - по аналогии с другими странами. Однако, строго говоря, постановка вопроса о том, как зарегистрировать медицинское изделие в ЕС, не вполне корректна. Процесс, который необходимо пройти, для того, чтобы ваша продукция могла продаваться в Европе, нельзя назвать регистрацией в привычном смысле слова, когда собирается регистрационное досье и подается в регуляторные органы для получения регистрационного удостоверения/сертификата и внесения регулятором в реестр зарегистрированной продукции.
Правильнее было бы говорить о поэтапном задокументированном приведении продукта и производителя в соответствие требованиям европейского законодательства, итогом которого является создание Декларации Соответствия (ее часто называют Декларацией соответствия CE или CE-Декларацией Соответствия) и нанесение CE-марки (ее часто называют CE-маркировкой). Последняя, в свою очередь, позволяет продавать медицинское изделие на всей территории ЕС.

Заинтересованы в рынке ЕС?
Свяжитесь с нами прямо сейчас:
+357 22253765
info@mdrc-consulting.com
Тем не менее поскольку формулировка «зарегистрировать медицинское изделие в ЕС» очень прочно вошла в обиход русскоязычных производителей медицинских изделий, мы будем использовать ее в тексте данной статьи, имея в виду весь процесс достижения соответствия регуляторным требованиям ЕС, начиная с классификации продукции и заканчивая нанесением CE-марки.
Законодательную основу для регистрации медицинских изделий в ЕС составляет Регламент Европейского Союза Medical Device Regulation 2017/745, известный также как MDR 2017/746 или просто MDR. Именно в нем вы найдете основные требования, предъявляемые в ЕС к производителям медицинских изделий и их продукции.
Помимо этого существует ряд спецификаций (Common Specifications), которые применимы к отдельным ситуациям, не описанным в MDR, а также ряд гармонизированных стандартов (например, ISO, EN и др.), на которые производитель может ориентироваться в процессе достижения соответствия требованиям MDR.
Ниже мы разберем какие этапы необходимо пройти и какие действия предпринять, чтобы добиться регуляторного соответствия MDR или иными словами, чтобы зарегистрировать медицинское изделие в ЕС.
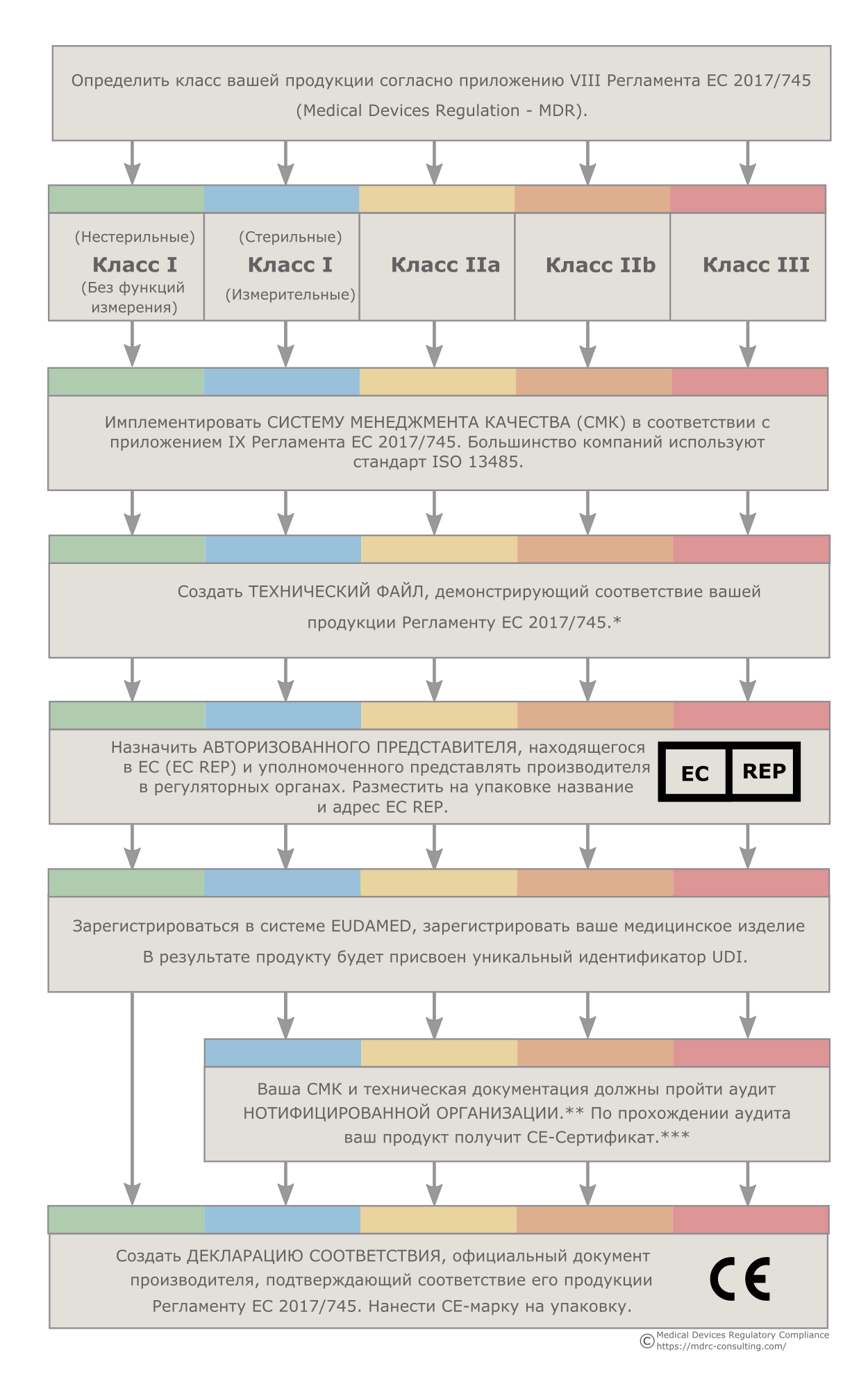
Определение класса риска изделия - первый шаг на пути регистрации медицинского продукта в ЕС. В ЕС используется классификация, основанная на правилах. Правила изложены в Приложении VIII MDR.
Классификация - это не только первый, но и критически важный этап регистрации медицинского изделия в ЕС. От установленного класса изделия зависит то, каковы будут дальнейшие процедуры регистрации. Поэтому перед тем как зарегистрировать медицинское изделие, производителю необходимо убедиться в том, что он правильно определил класс риска. Ошибка в определении класса может поставить под угрозу правильность прохождения последующих этапов и привести к большим временным и финансовым потерям. О том, как использовать правила классификации можно прочитать здесь (с примерами).
Каждый производитель медицинских изделий, продающий свою продукцию в Европейском Союзе, обязан иметь Систему Менеджмента Качества, отвечающую требованиям, изложенным в Приложении IX MDR. Причем географическое положение производителя не имеет значения, как и класс продукта. Если вы хотите зарегистрировать медицинское изделие в ЕС, у вас должна иметься адекватная СМК.
Одним из способов создания Системы Менеджмента Качества, отвечающей европейским требованиям, является внедрение в организации ISO 13485. Этот стандарт гармонизирован с требованиями MDR. Поэтому грамотно использовав его, вы добьетесь соответствия и европейскому законодательству.
Тем не менее соответствие ISO 13485 не является обязательным требованием при регистрации медицинских изделий в Европейском Союзе. Создать приемлемую СМК можно и опираясь на свои собственные внутренние стандарты. Особенно это относится к производителям медицинских изделий низких классов риска.
Если у вас уже имеется СМК, например, ISO 9001, вы можете использовать ее и адаптировать к требованиям MDR. Для этого, опять же, можно ориентироваться на ISO 13485 или на свои собственные разработки.
Технический файл продукта - это важнейший элемент всего процесса регистрации медицинского изделия в Европейском Союзе. Техническая документация должна демонстрировать безопасность и эффективность вашего продукта. Этот факт необходимо держать в уме, когда вы работаете над составлением документации.
Производитель имеет некоторую степень свободы при создании технических файлов. Тем не менее документация должна отвечать требованиям, изложенным в Приложениях II и III MDR, а технические файлы иметь структуру, представленную в тех же приложениях.

Доступ к техническим файлам предоставляется Авторизованным Представителям (см. ниже), Нотифицированным Организациям (см. Взаимодействие с Нотифицированной Организацией), а также надзорным органам Европейского Союза, если они решат проинспектировать вас.
Любая компания, находящаяся за пределами Европейского Союза, должна назначить Авторизованного Представителя - лицо или организацию, которая будет отвечать за продукцию производителя на рынке, обеспечивать репортирование нежелательных явлений, связанных с применением этой продукции, а также представлять производителя при взаимодействии с надзорными органами. Без Авторизованного Представителя неевропейской компании зарегистрировать медицинское изделие в ЕС невозможно.
Следует учитывать, что название Представителя должно будет наноситься на упаковку вашего продукта, вноситься в техническую документацию и регистрироваться в системе EUDAMED (см. ниже).
К выбору Авторизованного Представителя следует отнестись со всей серьезностью. Многие компании назначают Авторизованным Представителем одного из своих дистрибьюторов. Такой подход несет в себе определенные риски - если вы захотите разорвать отношения с дистрибьютором, вам придется искать нового Авторизованного Представителя, а значит менять маркировку продукта, вносить поправки в техническую документацию, уведомлять вашу нотифицированную организацию и корректировать данные в EUDAMED. Это, в свою очередь, может повлечь значительные потери.
Получение базового UDI-DI, уникального идентификационного кода, присваиваемого каждому медицинскому продукту, находящемуся на европейском рынке, обязательное требование, которое необходимо выполнить, чтобы зарегистрировать медицинское изделие в ЕС. Базовые UDI-DI выдаются производителям специальными организациями, аккредитованными Европейской Комиссией. Чтобы получить этот идентификатор, необходимо обратиться в одну из таких организаций. Более подробно о базовых UDI-DI можно прочитать здесь.
После того как у вас появится Авторизованный Представитель, вы сможете зарегистрировать вашу компанию и продукцию в электронной системе EUDAMED. В результате вам будет присвоен Регистрационный номер SRN, который станет идентификатором вашей компании на всей территории Европейского Союза.
SRN вам потребуется, в частности, при обращении в Нотифицированную Организацию, если такое обращение предусмотрено регуляторной процедурой, применимой к классу риска вашей продукции.
Если вы производите продукцию класса I, не являющейся стерильной и не используемой для измерений, то после регистрации в системе EUDAMED вы можете начинать наносить CE-марку на ваши медицинские изделия и продавать их в ЕС. Для всех остальных классов предусмотрен дополнительный контроль со стороны т.н. Нотифицированных Организаций.
Если вы хотите зарегистрировать в Европейском Союзе медицинское изделие III, IIb, IIa класса или стерильное или измерительное изделие I класса, то после регистрации в EUDAMED вы должны обратиться в одну из Нотифицированных Организаций Европейского Союза и предоставить ей доступ к документации вашей Системы Качества и к техническому файлу регистрируемого изделия. Нотифицированная Организация осуществит аудит вашей документации и, вполне возможно, проведет инспекцию ваших лабораторий и производственных помещений.
По итогам Нотифицированная Организация выдаст вашей компании сертификат соответствия требованиям MDR. Если это происходит, значит вам удалось зарегистрировать медицинское изделие в ЕС, вы можете наносить CE-марку и продавать ваш продукт на территории Европейского Союза.
Однако на этом процесс не заканчивается. Нотифицированная Организация назначает периодичность контрольных инспекций, необходимых для контроля поддержания Системы Качества, продукта и его документации в состоянии соответствия MDR. Чаще всего такие аудиты проводятся раз в год в течение первых нескольких лет, а затем их частота снижается.



Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.

+357 22253765
info@mdrc-consulting.com
Рынок медицинских изделий Европейского Союза строго регулируется. Если вы хотите продавать свою продукцию медицинского назначения в Европе, вам необходимо выполнить ряд требований, которые европейские регуляторы предъявляют к продуктам такого рода.
Иногда этот процесс называют регистрацией - по аналогии с другими странами. Однако, строго говоря, постановка вопроса о том, как зарегистрировать медицинское изделие в ЕС, не вполне корректна. Процесс, который необходимо пройти, для того, чтобы ваша продукция могла продаваться в Европе, нельзя назвать регистрацией в привычном смысле слова, когда собирается регистрационное досье и подается в регуляторные органы для получения регистрационного удостоверения/сертификата и внесения регулятором в реестр зарегистрированной продукции.
Правильнее было бы говорить о поэтапном задокументированном приведении продукта и производителя в соответствие требованиям европейского законодательства, итогом которого является создание Декларации Соответствия (ее часто называют Декларацией соответствия CE или CE-Декларацией Соответствия) и нанесение CE-марки (ее часто называют CE-маркировкой). Последняя, в свою очередь, позволяет продавать медицинское изделие на всей территории ЕС.
Заинтересованы в рынке ЕС?
Свяжитесь с нами прямо сейчас:
+357 22253765
info@mdrc-consulting.com
Тем не менее поскольку формулировка «зарегистрировать медицинское изделие в ЕС» очень прочно вошла в обиход русскоязычных производителей медицинских изделий, мы будем использовать ее в тексте данной статьи, имея в виду весь процесс достижения соответствия регуляторным требованиям ЕС, начиная с классификации продукции и заканчивая нанесением CE-марки.
Законодательную основу для регистрации медицинских изделий в ЕС составляет Регламент Европейского Союза Medical Device Regulation 2017/745, известный также как MDR 2017/746 или просто MDR. Именно в нем вы найдете основные требования, предъявляемые в ЕС к производителям медицинских изделий и их продукции.
Помимо этого существует ряд спецификаций (Common Specifications), которые применимы к отдельным ситуациям, не описанным в MDR, а также ряд гармонизированных стандартов (например, ISO, EN и др.), на которые производитель может ориентироваться в процессе достижения соответствия требованиям MDR.
Ниже мы разберем какие этапы необходимо пройти и какие действия предпринять, чтобы добиться регуляторного соответствия MDR или иными словами, чтобы зарегистрировать медицинское изделие в ЕС.
Определение класса риска изделия - первый шаг на пути регистрации медицинского продукта в ЕС. В ЕС используется классификация, основанная на правилах. Правила изложены в Приложении VIII MDR.
Классификация - это не только первый, но и критически важный этап регистрации медицинского изделия в ЕС. От установленного класса изделия зависит то, каковы будут дальнейшие процедуры регистрации. Поэтому перед тем как зарегистрировать медицинское изделие, производителю необходимо убедиться в том, что он правильно определил класс риска. Ошибка в определении класса может поставить под угрозу правильность прохождения последующих этапов и привести к большим временным и финансовым потерям. О том, как использовать правила классификации можно прочитать здесь (с примерами).
Каждый производитель медицинских изделий, продающий свою продукцию в Европейском Союзе, обязан иметь Систему Менеджмента Качества, отвечающую требованиям, изложенным в Приложении IX MDR. Причем географическое положение производителя не имеет значения, как и класс продукта. Если вы хотите зарегистрировать медицинское изделие в ЕС, у вас должна иметься адекватная СМК.
Одним из способов создания Системы Менеджмента Качества, отвечающей европейским требованиям, является внедрение в организации ISO 13485. Этот стандарт гармонизирован с требованиями MDR. Поэтому грамотно использовав его, вы добьетесь соответствия и европейскому законодательству.
Тем не менее соответствие ISO 13485 не является обязательным требованием при регистрации медицинских изделий в Европейском Союзе. Создать приемлемую СМК можно и опираясь на свои собственные внутренние стандарты. Особенно это относится к производителям медицинских изделий низких классов риска.
Если у вас уже имеется СМК, например, ISO 9001, вы можете использовать ее и адаптировать к требованиям MDR. Для этого, опять же, можно ориентироваться на ISO 13485 или на свои собственные разработки.
Технический файл продукта - это важнейший элемент всего процесса регистрации медицинского изделия в Европейском Союзе. Техническая документация должна демонстрировать безопасность и эффективность вашего продукта. Этот факт необходимо держать в уме, когда вы работаете над составлением документации.
Производитель имеет некоторую степень свободы при создании технических файлов. Тем не менее документация должна отвечать требованиям, изложенным в Приложениях II и III MDR, а технические файлы иметь структуру, представленную в тех же приложениях.
Доступ к техническим файлам предоставляется Авторизованным Представителям (см. ниже), Нотифицированным Организациям (см. Взаимодействие с Нотифицированной Организацией), а также надзорным органам Европейского Союза, если они решат проинспектировать вас.
Любая компания, находящаяся за пределами Европейского Союза, должна назначить Авторизованного Представителя - лицо или организацию, которая будет отвечать за продукцию производителя на рынке, обеспечивать репортирование нежелательных явлений, связанных с применением этой продукции, а также представлять производителя при взаимодействии с надзорными органами. Без Авторизованного Представителя неевропейской компании зарегистрировать медицинское изделие в ЕС невозможно.
Следует учитывать, что название Представителя должно будет наноситься на упаковку вашего продукта, вноситься в техническую документацию и регистрироваться в системе EUDAMED (см. ниже).
К выбору Авторизованного Представителя следует отнестись со всей серьезностью. Многие компании назначают Авторизованным Представителем одного из своих дистрибьюторов. Такой подход несет в себе определенные риски - если вы захотите разорвать отношения с дистрибьютором, вам придется искать нового Авторизованного Представителя, а значит менять маркировку продукта, вносить поправки в техническую документацию, уведомлять вашу нотифицированную организацию и корректировать данные в EUDAMED. Это, в свою очередь, может повлечь значительные потери.
Получение базового UDI-DI, уникального идентификационного кода, присваиваемого каждому медицинскому продукту, находящемуся на европейском рынке, обязательное требование, которое необходимо выполнить, чтобы зарегистрировать медицинское изделие в ЕС. Базовые UDI-DI выдаются производителям специальными организациями, аккредитованными Европейской Комиссией. Чтобы получить этот идентификатор, необходимо обратиться в одну из таких организаций. Более подробно о базовых UDI-DI можно прочитать здесь.
После того как у вас появится Авторизованный Представитель, вы сможете зарегистрировать вашу компанию и продукцию в электронной системе EUDAMED. В результате вам будет присвоен Регистрационный номер SRN, который станет идентификатором вашей компании на всей территории Европейского Союза.
SRN вам потребуется, в частности, при обращении в Нотифицированную Организацию, если такое обращение предусмотрено регуляторной процедурой, применимой к классу риска вашей продукции.
Если вы производите продукцию класса I, не являющейся стерильной и не используемой для измерений, то после регистрации в системе EUDAMED вы можете начинать наносить CE-марку на ваши медицинские изделия и продавать их в ЕС. Для всех остальных классов предусмотрен дополнительный контроль со стороны т.н. Нотифицированных Организаций.
Если вы хотите зарегистрировать в Европейском Союзе медицинское изделие III, IIb, IIa класса или стерильное или измерительное изделие I класса, то после регистрации в EUDAMED вы должны обратиться в одну из Нотифицированных Организаций Европейского Союза и предоставить ей доступ к документации вашей Системы Качества и к техническому файлу регистрируемого изделия. Нотифицированная Организация осуществит аудит вашей документации и, вполне возможно, проведет инспекцию ваших лабораторий и производственных помещений.
По итогам Нотифицированная Организация выдаст вашей компании сертификат соответствия требованиям MDR. Если это происходит, значит вам удалось зарегистрировать медицинское изделие в ЕС, вы можете наносить CE-марку и продавать ваш продукт на территории Европейского Союза.
Однако на этом процесс не заканчивается. Нотифицированная Организация назначает периодичность контрольных инспекций, необходимых для контроля поддержания Системы Качества, продукта и его документации в состоянии соответствия MDR. Чаще всего такие аудиты проводятся раз в год в течение первых нескольких лет, а затем их частота снижается.
Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.
+357 22253765
info@mdrc-consulting.com
Мы будем рады обсудить ваш новый проект!