С 26 мая 2022 года все производители изделий для in vitro диагностики, продающие свою продукцию в Европе, должны обеспечить соответствие своей продукции европейскому регламенту IVDR 2017/746. Ниже речь пойдет о том, что это значит на практике, и какие действия следует предпринять производителям, чтобы соблюсти требования регуляторных властей Европейского Союза, связанные с переходом на новое законодательство.
Для европейских регуляторов изменение законодательства ЕС в области продукции для in vitro диагностики - давно назревшая необходимость. В Директиве ЕС IVDD 98/79 имелся целый ряд дефицитов, которые новый регламент должен устранить. Кроме того, введение в силу IVDR позволяет в значительной степени гармонизировать регуляторные требования Европейского Союза с таковыми других стран, в первую очередь с требованиями FDA.

Заинтересованы в рынке ЕС?
Свяжитесь с нами прямо сейчас:
+357 22253765
info@mdrc-consulting.com
В целом, с точки зрения европейских регуляторных властей переход с IVDD на IVDR несет в себе следующие позитивные изменения:
В то же время с точки зрения производителей медицинских изделий для in vitro диагностики переход с IVDD на IVDR означает дополнительную регуляторную нагрузку, а значит дополнительные риски и затраты. И главные из них связаны с введением новой классификации изделий для in vitro диагностики и возникновением необходимости привлечения нотифицированных организаций к оценке регуляторного соответствия для тех продуктов, для которых раньше этого не требовалось.
Повышение роли нотифицированных организаций и возникновение потребности в сертификации изделий и производителя можно представить следующим образом:

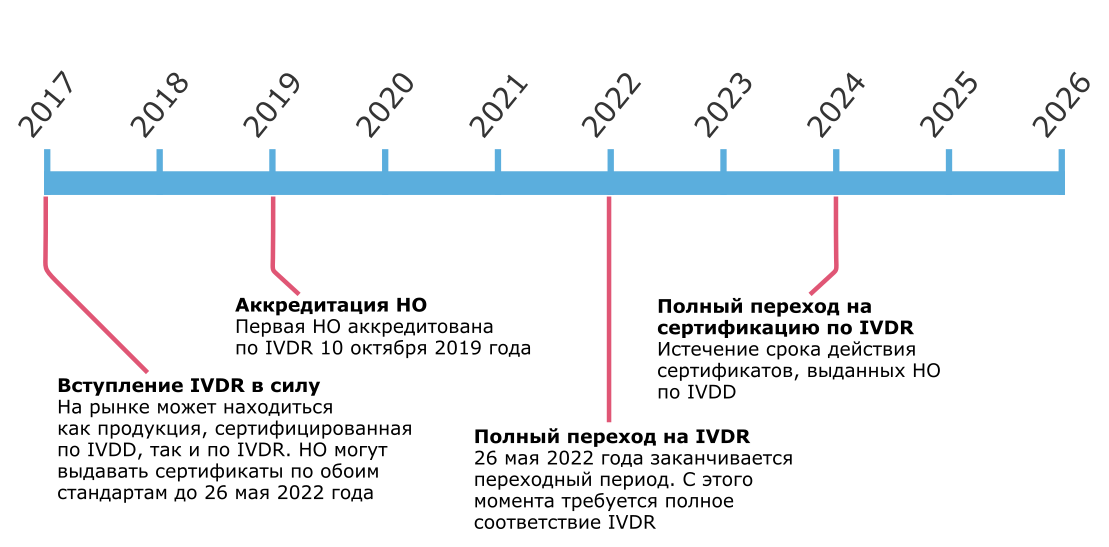
Регламент IVDR 2017/746 вступил в силу 26 мая 2017 года. С этого момента начался пятилетний переходный период, в течение которого для продукции для in vitro диагностики действовало и старое, и новое законодательство. С 26 мая 2022 года Директива ЕС 93/42 больше не действует, и все производители должны отвечать требованиям IVDR.
IVDR не следует рассматривать просто как некое продолжение IVDD. Введение этого регламента предполагает концептуально новый подход ко многим аспектам регулирования обращения медицинских изделий для in vitro диагностики в Европейском Союзе. Наиболее существенные изменения приводятся ниже:
IVDR дает более четкие определения ключевым понятиям и охватывает более широкий спектр продуктов, по сравнению с IVDD, включая изделия, в которых используются новые технологии, например, программную продукцию или изделия для персонифицированной диагностики. Кроме того, новый регламент регулирует диагностические услуги предоставляемые с помощью сети Интернет с использованием диагностической продукции, находящейся за пределами ЕС (например, дистанционное генетические тестирование).
Одним из главных изменений, связанных с вступлением в силу IVDR, стало введение классификации, основанной на правилах, в отличие от классификации, основанной на списках, представленной в IVDD.
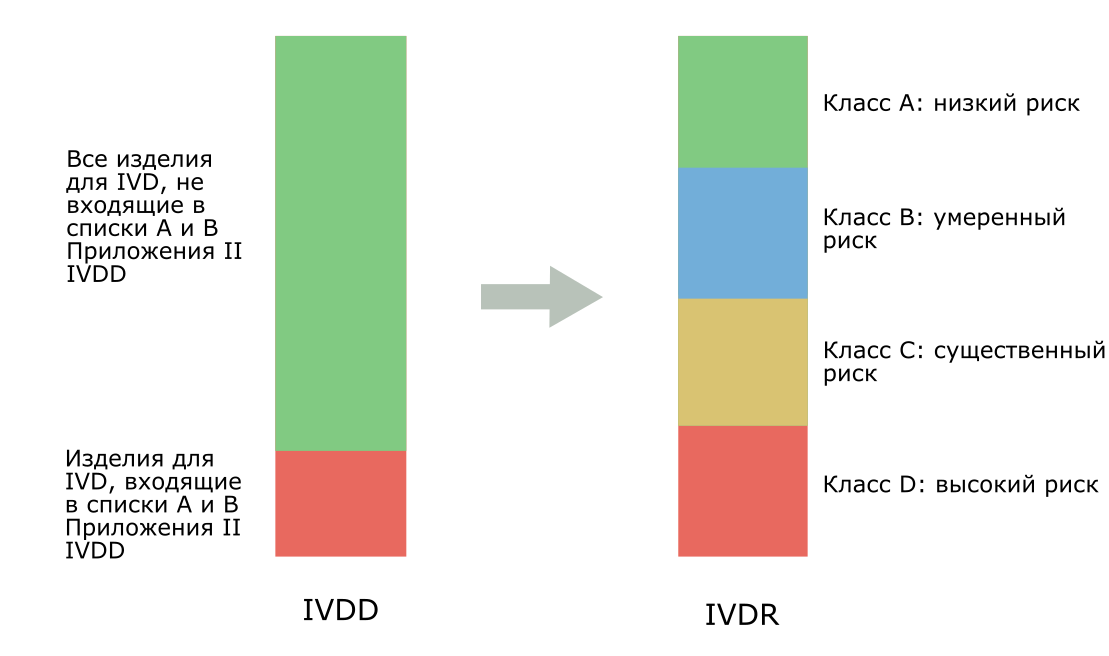
По старой классификации, медицинские изделия для in vitro диагностики делились на «общие» изделия и изделия, относящиеся к спискам А или В Приложения II IVDD. Привлечения нотифицированной организации и сертификации требовали только изделия, относящиеся к указанным спискам (они составляли около 20% всех продуктов для IVD).
По новой классификации медицинские изделия для in vitro диагностики делятся на четыре класса по степени риска, связанного с их применением - классы A, B, C и D. Привлечения нотифицированных организаций требуют все продукты, кроме относящихся к классу А, т.е. 80% всех изделий для in vitro диагностики. Отсюда и возрастание необходимости привлечения нотифицированных организаций к оценке регуляторного соответствия (см. рис. 1).
Старую и новую системы классификации можно представить следующим образом:
Регламент ЕС IVDR 2017/746 требует от производителя более скрупулезного подхода в управлении жизненным циклом продукции, в том числе:
Новый регламент ЕС возлагает на экономических операторов (производителей, импортеров, дистрибьюторов, авторизованных представителей) дополнительную ответственность в отношении контроля цепей поставок. Согласно IVDR каждый участник цепи обязан контролировать регуляторное соответствие предыдущих звеньев цепи, с которыми он непосредственно работает. Также, IVDR дает нотифицированным организациям право инспектировать не только производителя, но и его поставщиков и субконтракторов.
Новым требованием является наличие у производителей и авторизованных представителей т.н. лица, отвечающего за регуляторное соответствие (PRRC - Person Responsible for Regulatory Compliance). Такое лицо должно либо находиться в штате компании, либо (если компания относится к малым или микропредприятиям) может выполнять свои обязанности, находясь вне штата - на контрактной основе. PRRC отвечает не только за выполнение требований IVDR компанией, для которой он работает, но и за регуляторное соответствие всей цепи поставок.
Изменение европейского законодательства, касающегося медицинских изделий для in vitro диагностики требует от производителя целого ряда мер. Среди них - пересмотр регуляторной стратегии, классификация продукции согласно требованиям IVDR, gap-анализ технической документации и системы качества, поиск нотифицированной организации (при необходимости) и др.
Грамотные действия, направленные на своевременный переход на новое законодательство позволят производителю избежать потерь и своевременно обеспечить регуляторное соответствие требованиям европейского рынка.



Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.

+357 22253765
info@mdrc-consulting.com
С 26 мая 2022 года все производители изделий для in vitro диагностики, продающие свою продукцию в Европе, должны обеспечить соответствие своей продукции европейскому регламенту IVDR 2017/746. Ниже речь пойдет о том, что это значит на практике, и какие действия следует предпринять производителям, чтобы соблюсти требования регуляторных властей Европейского Союза, связанные с переходом на новое законодательство.
Для европейских регуляторов изменение законодательства ЕС в области продукции для in vitro диагностики - давно назревшая необходимость. В Директиве ЕС IVDD 98/79 имелся целый ряд дефицитов, которые новый регламент должен устранить. Кроме того, введение в силу IVDR позволяет в значительной степени гармонизировать регуляторные требования Европейского Союза с таковыми других стран, в первую очередь с требованиями FDA.
Заинтересованы в рынке ЕС?
Свяжитесь с нами прямо сейчас:
+357 22253765
info@mdrc-consulting.com
В целом, с точки зрения европейских регуляторных властей переход с IVDD на IVDR несет в себе следующие позитивные изменения:
В то же время с точки зрения производителей медицинских изделий для in vitro диагностики переход с IVDD на IVDR означает дополнительную регуляторную нагрузку, а значит дополнительные риски и затраты. И главные из них связаны с введением новой классификации изделий для in vitro диагностики и возникновением необходимости привлечения нотифицированных организаций к оценке регуляторного соответствия для тех продуктов, для которых раньше этого не требовалось.
Повышение роли нотифицированных организаций и возникновение потребности в сертификации изделий и производителя можно представить следующим образом:
Регламент IVDR 2017/746 вступил в силу 26 мая 2017 года. С этого момента начался пятилетний переходный период, в течение которого для продукции для in vitro диагностики действовало и старое, и новое законодательство. С 26 мая 2022 года Директива ЕС 93/42 больше не действует, и все производители должны отвечать требованиям IVDR.
IVDR не следует рассматривать просто как некое продолжение IVDD. Введение этого регламента предполагает концептуально новый подход ко многим аспектам регулирования обращения медицинских изделий для in vitro диагностики в Европейском Союзе. Наиболее существенные изменения приводятся ниже:
IVDR дает более четкие определения ключевым понятиям и охватывает более широкий спектр продуктов, по сравнению с IVDD, включая изделия, в которых используются новые технологии, например, программную продукцию или изделия для персонифицированной диагностики. Кроме того, новый регламент регулирует диагностические услуги предоставляемые с помощью сети Интернет с использованием диагностической продукции, находящейся за пределами ЕС (например, дистанционное генетические тестирование).
Одним из главных изменений, связанных с вступлением в силу IVDR, стало введение классификации, основанной на правилах, в отличие от классификации, основанной на списках, представленной в IVDD.
По старой классификации, медицинские изделия для in vitro диагностики делились на «общие» изделия и изделия, относящиеся к спискам А или В Приложения II IVDD. Привлечения нотифицированной организации и сертификации требовали только изделия, относящиеся к указанным спискам (они составляли около 20% всех продуктов для IVD).
По новой классификации медицинские изделия для in vitro диагностики делятся на четыре класса по степени риска, связанного с их применением - классы A, B, C и D. Привлечения нотифицированных организаций требуют все продукты, кроме относящихся к классу А, т.е. 80% всех изделий для in vitro диагностики. Отсюда и возрастание необходимости привлечения нотифицированных организаций к оценке регуляторного соответствия (см. рис. 1).
Старую и новую системы классификации можно представить следующим образом:
Регламент ЕС IVDR 2017/746 требует от производителя более скрупулезного подхода в управлении жизненным циклом продукции, в том числе:
Новый регламент ЕС возлагает на экономических операторов (производителей, импортеров, дистрибьюторов, авторизованных представителей) дополнительную ответственность в отношении контроля цепей поставок. Согласно IVDR каждый участник цепи обязан контролировать регуляторное соответствие предыдущих звеньев цепи, с которыми он непосредственно работает. Также, IVDR дает нотифицированным организациям право инспектировать не только производителя, но и его поставщиков и субконтракторов.
Новым требованием является наличие у производителей и авторизованных представителей т.н. лица, отвечающего за регуляторное соответствие (PRRC - Person Responsible for Regulatory Compliance). Такое лицо должно либо находиться в штате компании, либо (если компания относится к малым или микропредприятиям) может выполнять свои обязанности, находясь вне штата - на контрактной основе. PRRC отвечает не только за выполнение требований IVDR компанией, для которой он работает, но и за регуляторное соответствие всей цепи поставок.
Изменение европейского законодательства, касающегося медицинских изделий для in vitro диагностики требует от производителя целого ряда мер. Среди них - пересмотр регуляторной стратегии, классификация продукции согласно требованиям IVDR, gap-анализ технической документации и системы качества, поиск нотифицированной организации (при необходимости) и др.
Грамотные действия, направленные на своевременный переход на новое законодательство позволят производителю избежать потерь и своевременно обеспечить регуляторное соответствие требованиям европейского рынка.
Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.
+357 22253765
info@mdrc-consulting.com
Мы будем рады обсудить ваш новый проект!