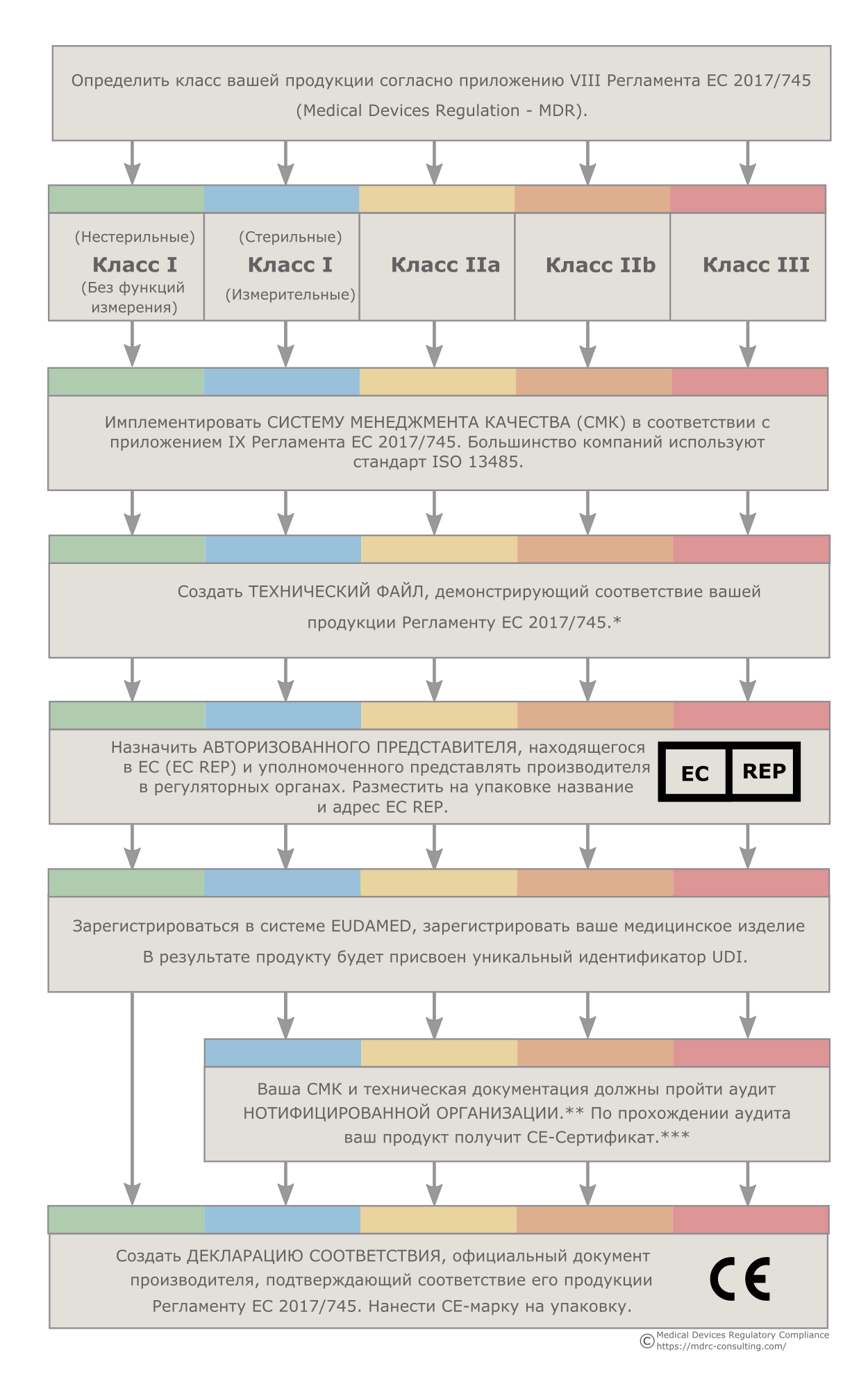
*Продукция классов IIb и III с большой вероятностью потребует большого количества клинических данных. В ряде случаев могут использоваться существующие научные данные, но нередко требуется проведение клинических исследований. Клинические исследования, проводимые в ЕС должны быть одобрены Европейскими регуляторными агентствами. Планы и отчеты по клиническим испытаниям помещаются в соответствующий раздел технической документации.
**Нотифицированная Организация - организация, аккредитованная Европейским Союзом на контроль над производителями продукции медицинского назначения.
***CE-Сертификат не выдается продуктам класса I (нестерильным, без функции измерения), поскольку для медицинских изделий данного класса соответствие требованиям Регламента ЕС 2017/745 заявляется производителем в порядке самодекларации.
MDRC помогает производителям изделий медицинского назначения достичь соответствия регуляторным требованиям Европейского Союза, зарегистрировать и вывести на европейский рынок их продукцию.
В ЕС определение класса изделий медицинского назначения осуществляется на основе ряда правил, изложенных в приложении VIII Регламента ЕС 2017/745. Поэтому медицинский продукт определенного класса по классификации РФ, США, Китая или другой страны, не входящей в Европейский Союз, в ЕС может относиться к другому классу продукции. Правильное определение класса вашей продукции является критически важным, поскольку от класса будет зависеть то, каким образом будет осуществляться регистрация и вывод на рынок ваших продуктов. Мы поможем вам правильно классифицировать вашу продукцию.
Наличие технического файла является обязательным требованием ЕС, независимо от класса и типа продукта. Досье разработки обязательны для продуктов класса III. Досье разработки отличаются от технических файлов рядом дополнительных требований - в частности необходимостью обширной клинической программы. Мы создали многочисленные технические файлы и досье разработки для целого ряда различных продуктов и готовы помочь вам в создании технической и клинической документации для вашей продукции. Кроме того, мы проводим клиническую оценку продукции, разрабатываем инструкции по применению изделий медицинского назначения и помогаем добиться соответствия ISO 14971.
У MDRC имеются готовые процедуры и шаблоны документов, которые соответствуют регуляторным требованиям большинства стран и которые могут быть имплементированы в любой компании. Большинство компаний предпочитают использовать стандарт ISO 13485. Мы готовы помочь вам с внедрением СМК. Если у вас уже имеется СМК, мы поможем вам добиться ее соответствия европейским требованиям.

MDR (EU 2017/745) и IVDR (EU 2017/746) требуют наличия у производителя медицинских изделий PRRC (Person Responsible for Regulatory Compliance).
Компании, находящиеся вне Европейского Союза, должны назначить Авторизованного Представителя в ЕС (EC REP), который будет представлять их в регуляторных органах ЕС. Хотя эту роль может выполнять дистрибьютор, наличие независимого Авторизованного Представителя несет в себе большие преимущества, поскольку позволяет менять дистрибьюторов в любое удобное для вас время. Наше германское представительство готово взять на себя роль Авторизованного Представителя для вашей компании.
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, должны ежегодно проходить аудит Нотифицированной Организации. Мы поможем вам подготовиться к аудиту. Помимо этого мы можем провести для ваших работников тренинги по процедурам получения CE-марки, Регламенту ЕС 2017/745, а также внутреннему аудиту.
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, взаимодействуют с регуляторными оранами ЕС через Нотифицированные Организации. За поиск и выбор Нотифицированной организации, а также коммуникацию с ней отвечает сам производитель. Мы имеем большой опыт работы с Нотифицированными Организациями из разных стран ЕС и готовы помочь вам выбрать подходящую организацию и наладить взаимодействие с ней.
Согласно Регламенту ЕС дистрибьюторы изделий медицинского назначения в ЕС должны отвечать целому ряду регуляторных требований. Далеко не каждая компания, готовая продавать вашу продукцию, может стать вашим дистрибьютором. В настоящий момент в Европейском Союзе - 27 стран и более 20 официальных языков. Оценка и квалификация дистрибьюторов в таких условиях - непростая задача. Мы поможем вам найти, проанализировать и отобрать подходящих дистрибьюторов.
*Продукция классов IIb и III с большой вероятностью потребует большого количества клинических данных. В ряде случаев могут использоваться существующие научные данные, но нередко требуется проведение клинических исследований. Клинические исследования, проводимые в ЕС должны быть одобрены Европейскими регуляторными агентствами. Планы и отчеты по клиническим испытаниям помещаются в соответствующий раздел технической документации.
**Нотифицированная Организация - организация, аккредитованная Европейским Союзом на контроль над производителями продукции медицинского назначения.
***CE-Сертификат не выдается продуктам класса I (нестерильным, без функции измерения), поскольку для медицинских изделий данного класса соответствие требованиям Регламента ЕС 2017/745 заявляется производителем в порядке самодекларации.
MDRC помогает производителям изделий медицинского назначения достичь соответствия регуляторным требованиям Европейского Союза, зарегистрировать и вывести на европейский рынок их продукцию.
В ЕС определение класса изделий медицинского назначения осуществляется на основе ряда правил, изложенных в приложении VIII Регламента ЕС 2017/745. Поэтому медицинский продукт определенного класса по классификации РФ, США, Китая или другой страны, не входящей в Европейский Союз, в ЕС может относиться к другому классу продукции. Правильное определение класса вашей продукции является критически важным, поскольку от класса будет зависеть то, каким образом будет осуществляться регистрация и вывод на рынок ваших продуктов. Мы поможем вам правильно классифицировать вашу продукцию.
Наличие технического файла является обязательным требованием ЕС, независимо от класса и типа продукта. Досье разработки обязательны для продуктов класса III. Досье разработки отличаются от технических файлов рядом дополнительных требований - в частности необходимостью обширной клинической программы. Мы создали многочисленные технические файлы и досье разработки для целого ряда различных продуктов и готовы помочь вам в создании технической и клинической документации для вашей продукции. Кроме того, мы проводим клиническую оценку продукции, разрабатываем инструкции по применению изделий медицинского назначения и помогаем добиться соответствия ISO 14971.
У MDRC имеются готовые процедуры и шаблоны документов, которые соответствуют регуляторным требованиям большинства стран и которые могут быть имплементированы в любой компании. Большинство компаний предпочитают использовать стандарт ISO 13485. Мы готовы помочь вам с внедрением СМК. Если у вас уже имеется СМК, мы поможем вам добиться ее соответствия европейским требованиям.
Компании, находящиеся вне Европейского Союза, должны назначить Авторизованного Представителя в ЕС (EC REP), который будет представлять их в регуляторных органах ЕС. Хотя эту роль может выполнять дистрибьютор, наличие независимого Авторизованного Представителя несет в себе большие преимущества, поскольку позволяет менять дистрибьюторов в любое удобное для вас время. Наше германское представительство готово взять на себя роль Авторизованного Представителя для вашей компании.
MDR (EU 2017/745) и IVDR (EU 2017/746) требуют наличия у производителя медицинских изделий PRRC (Person Responsible for Regulatory Compliance).
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, должны ежегодно проходить аудит Нотифицированной Организации. Мы поможем вам подготовиться к аудиту. Помимо этого мы можем провести для ваших работников тренинги по процедурам получения CE-марки, Регламенту ЕС 2017/745, а также внутреннему аудиту.
Производители медицинских изделий классов IIa, IIb и III, а также стерильных продуктов класса I или изделий класса I, предназначенных для измерений, взаимодействуют с регуляторными оранами ЕС через Нотифицированные Организации. За поиск и выбор Нотифицированной организации, а также коммуникацию с ней отвечает сам производитель. Мы имеем большой опыт работы с Нотифицированными Организациями из разных стран ЕС и готовы помочь вам выбрать подходящую организацию и наладить взаимодействие с ней.
Согласно Регламенту ЕС дистрибьюторы изделий медицинского назначения в ЕС должны отвечать целому ряду регуляторных требований. Далеко не каждая компания, готовая продавать вашу продукцию, может стать вашим дистрибьютором. В настоящий момент в Европейском Союзе - 27 стран и более 20 официальных языков. Оценка и квалификация дистрибьюторов в таких условиях - непростая задача. Мы поможем вам найти, проанализировать и отобрать подходящих дистрибьюторов.