Европейский Союз и США - два крупнейших рынка медицинских изделий. Ни один производитель не откажется вывести свою продукцию на один из них - а еще лучше на оба. Но, как известно, эти рынки строго регулируются. А значит, чтобы на них попасть, необходимо пройти определенные регуляторные процедуры.
Логично предположить, что разные медицинские изделия требуют несколько разных подходов. Было бы странно, если бы, скажем, к эластичному бинту и к вентрикулоперитонеальному шунту предъявлялись одинаковые требования. И здесь нам на помощь приходит классификация медицинских изделий, которая и определяет регуляторные пути, которыми придется пройти вашему продукту, прежде чем он окажется на рынке и принесет вашей компании миллионы.
Для того чтобы понять, какова будет процедура получения одобрения вашего продукта (а значит каковы будут сроки одобрения и каких затрат потребует этот процесс), необходимо определить, к какому классу риска он относится. О том, как это сделать, и пойдет речь в данной брошюре.

Скачать эту статью в PDF-формате
Здесь следует заметить, что американский и европейский подходы весьма существенно различаются. Мы разберем оба этих подхода и рассмотрим две классификации на конкретных примерах медицинских изделий. Но сначала давайте разберемся с важнейшим вопросом, над которым, почему-то, производители медицинских изделий стараются не задумываться. А именно: на каком этапе следует обращаться к теме классификации?
Многие компании, разрабатывающие медицинскую продукцию, откладывают определение класса продукта на конец разработки. Мол, не будем городить огород - сначала создадим изделие, а там уж определим, каким именно образом мы будем выводить его на рынок. Ответственно заявляем: это плохой подход!
Вам следует определить класс изделия как можно раньше - желательно в самом начале разработки, на этапе создания идеи, а затем контролировать возможные изменения класса продукта. Дело в том, что, если вы этого не сделаете, то в итоге вы можете получить изделие более высокого класса риска, чем вам представлялось вначале. А временные и финансовые затраты на регистрацию продукта по мере увеличения класса риска возрастают в геометрической прогрессии.
В результате вместо трех-четырех месяцев и нескольких тысяч долларов получение одобрения может потребовать пару лет и затрат, измеряемых сотнями тысяч. Вполне возможно, что компания окажется не готова к таким радикальным изменениям плана и бюджета разработки. И в итоге продукт, который мог бы продаваться в Европе и США и приносить компании колоссальные прибыли, навсегда остается на локальном рынке. А то и вовсе разработка будет остановлена.
Мораль проста: классификация медицинского изделия - это такой же управляемый элемент проекта разработки, как таймлайны, бюджет, качество или человеческие ресурсы. Но для того, чтобы управлять классификацией, вам необходимо определить класс будущего изделия как можно раньше и соответствующим образом планировать ресурсы.
Все это не значит, что в процессе разработки класс не может меняться. Вполне возможно, что класс финального продукта будет отличаться от класса, определенного исходно. Но это будет осознанное решение, принятое с учетом оценки проектных рисков и необходимости пересмотра ресурсов.
Как мы уже упомянули, регуляторные органы ЕС и США по-разному подходят к вопросам классификации медицинских изделий. Тем не менее между классами изделий можно провести аналогию.
| Низкий риск | Средний риск | Высокий риск | ||
|---|---|---|---|---|
| ЕС | I | IIa | IIb | III |
| США | I | II | III | |
Европейская классификация основана на правилах. Правила классификации приводятся в нормативном документе ЕС, известном как Medical Device Regulation (MDR), он же Регламент ЕС 2017/745 (Regulation (EU) 2017/745). Данный документ находится в свободном доступе.
Пояснение: В данной статье мы не рассматриваем европейское законодательство, касающееся изделий для in vitro диагностики. Правила классификации для продукции in vitro изложены в нормативном документе In Vitro Medical Device Regulation (IVDR) (Regualtion 2017/746).
Всего таких правил - 22. Чтобы определить класс вашего продукта, вам необходимо проверить его соответствие каждому из правил, начиная с Правила 1. Когда вы пройдете все применимые к вашему продукту правила, вам должно стать понятно, к какому классу относится ваш продукт.
Правила разбиты на 4 группы. Первая группа относится к неинвазивным изделиям. Вторая - к инвазивным. Третья - к активным (т.е. приводимым в движение каким-либо источником энергии). Четвертая - специальные правила, применимые к отдельным категориям продукции.
В отличие от европейских регуляторов (европейская система децентрализована, поэтому говорить о едином европейском регуляторе было бы некорректно), FDA определяет класс каждого изделия и хранит его индивидуальные данные в гигантской базе данных (Принципы классификации изложены в нормативе 21 CFR Part 860). Чтобы определить класс вашего изделия, вам необходимо обратиться к этой базе данных (доступ к ней имеет любой пользователь интернета) и найти продукт, аналогичный вашему. Класс вашего продукта будет соответствовать классу аналога. Звучит просто. Однако на практике все выглядит несколько сложнее.
Проблема в том, что база данных FDA действительно гигантская, и, чтобы найти в ней аналог вашего изделия, нужно использовать правильные поисковые слова. Поэтому определение класса медицинского изделия в США начинается с составления списка поисковых слов. При этом вам необходимо учитывать правильность написания и особенности американской терминологии, которая может отличаться от терминологии, к которой привыкли европейцы.
Чтобы получить представление о том, как определяется класс медицинского изделия в Европейском Союзе, обратимся к примерам двух продуктов. Пусть оба продукта относятся к области ортопедии: хирургическое сверло и ортопедическая стелька. Проверим оба продукта на соответствие правилам европейской классификации, изложенным в Приложении VIII Регламента ЕС 2017/745 (чтобы лучше понять то, о чем идет речь ниже, хорошо иметь текст приложения перед глазами).
Первые четыре правила относятся только к неинвазивным изделиям. Хирургическое сверло - инвазивное изделие. Поэтому для определения класса следует сразу перейти к разделу "Инвазивные изделия", который начинается с Правила 5.

Правило 5 относится к инвазивным изделиям, проникающим в тело человека через естественные отверстия. Поэтому к сверлу оно также не применимо, поскольку добраться до кости, не нарушив целостности тканей невозможно. В свою очередь, Правило 6 относится к хирургическим инвазивным (нарушающим целостность тканей) изделиям, значит, сверло подпадает под действие этого правила.
Правило 6 гласит: все хирургические инвазивные изделия, применяемые краткосрочно (менее 60 минут) относятся к классу IIa. Соответственно, сверло должно было бы относиться к классу IIa. Однако у Правила 6 имеются оговорки, которые приводятся в виде списка под текстом правила. Одна из оговорок гласит, что все многоразовые хирургические инструменты относятся к классу I.
Это хорошая новость для производителя, но надо проверить изделие на соответствие и другим правилам. После проверки мы устанавливаем, что хирургическое сверло не подпадает под действие других правил. Соответственно данный продукт относится к классу I.
Изделия класса I делятся на обычный класс I, а также на стерильные изделия класса I и изделия класса I с измерительными функциями. Регуляторные процедуры для двух последних категорий несколько сложнее и строже.
Сверло не имеет измерительной функции - из этой категории мы можем его сразу исключить. Что касается стерильности - безусловно в условиях операционной сверло используется в стерильном состоянии. Но для классификации важно не это, а то, в каком состоянии оно поставляется покупателям.
Многоразовое сверло поставляется в нестерильном состоянии. Поэтому в категорию стерильных изделий класса I оно не попадает. В итоге получаем изделие, относящееся к самому низкому классу риска - класс I (нестерильные, без функции измерения).
Теперь определим класс ортопедической стельки. Это неинвазивное изделие. Соответственно, классификацию начинаем с раздела «Неинвазивные изделия», т.е. с Правила 1.

Правило 1 гласит: Все неинвазивные изделия относятся к классу I, если все дальнейшие правила неприменимы к ним. Проверяем все дальнейшие правила и убеждаемся, что к ортопедической стельке они неприменимы. Следовательно, стелька относится к классу I. Поскольку она не используется для измерений и не поставляется в стерильном виде, получаем класс 1 (нестерильные, без функции измерения).
Таким образом, и хирургическое сверло, и ортопедическая стелька относятся к классу I (нестерильные, без функции измерения). Регуляторная процедура для таких изделий будет самая простая. Фактически такой продукт может выводиться на рынок без разрешения регуляторов (нотифицированных организаций).
Тем не менее мы пришли к подобной классификации двух изделий несколько разными путями. Для хирургического сверла определяющим стало Правило 5, для ортопедической стельки - Правило 1. В соответствующем разделе технического файла класс изделия приводится с указанием определяющего правила.
Например:
Device: Drill bit
Class I (Rule 5)
Device: Orthotic shoe insert
Class I (Rule 1).
Теперь рассмотрим классификацию медицинских изделий в США. К какому классу будут относиться, скажем, костные кусачки. Для того чтобы определить класс, нам потребуется найти продукт-аналог (predicate device), который был ранее зарегистрирован FDA и находится в базе данных регулятора.
Для этого загружаем базу данных и осуществляем поиск по запросу "wire cutter". В итоге находим ранее зарегистрированный продукт, который может выступать в роли аналога.
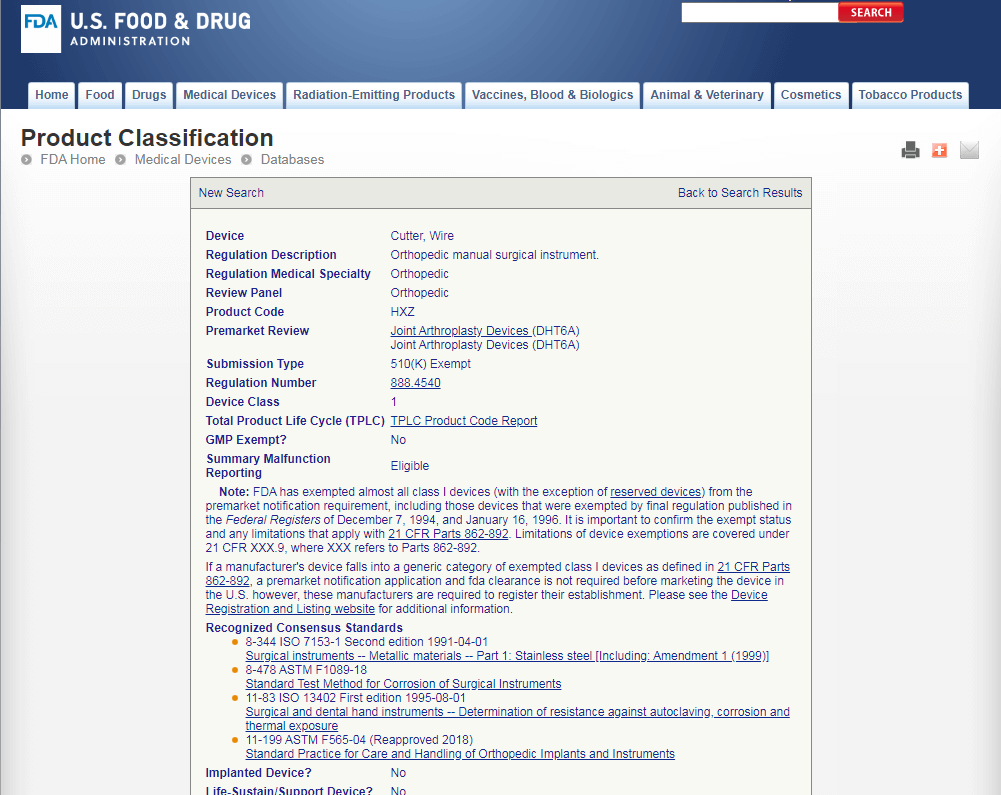
Продукт-аналог относится к классу I. Значит и ваш продукт относится к классу I. В базе данных FDA содержится и другая полезная информация о продукте-аналоге. Пройдя по ссылкам, приведенным в результатах поиска можно получить подробное описание продукта или его дженериковой группы, а также данные о проблемах с продуктом, отзывах и т.д. Изучив информацию, касающуюся продукта-аналога, можно установить является ли ваш продукт исключением из 510(k), а также должен ли он непременно производиться в условиях GMP или, опять же, является исключением.
Итак, полная классификация костных кусачек в США будет выглядеть следующим образом:
Device: Cutter, Wire
Class I. Product Code: HTW. Regulation No.: 888.4540
Аналогичным образом осуществляется классификация любого медицинского изделия. Например, уже знакомого нам хирургического сверла.

Как правило, поиск выдает несколько продуктов, которые могли бы выступить в роли аналога для вашего изделия. Выбор продукта-аналога осуществляется самим производителем, который отвечает за правильность этого выбора. При этом производитель должен руководствоваться характеристиками своего изделия и продуктов-аналогов.
Мы рассмотрели примеры изделий, относящихся к классу I и в ЕС, и в США. Тем не менее классы изделий в двух данных юрисдикциях совпадают далеко не всегда. Европейские классы IIа и IIb совсем необязательно соответствуют американскому классу II. Точно так же могут не совпадать и классы I и III. Рассмотрим пример эндопротеза тазобедренного сустава.

Правило 8 европейской классификации гласит: Все имплантируемые изделия, а также хирургические инвазивные изделия для долгосрочного применения относятся к классу IIb...
Далее приводится ряд оговорок, среди которых есть и такая: если только они не являются частичными или полными заменителями суставов. В последнем случае они относятся к классу III (за исключением винтов, клиньев, пластин). Соответственно, классификация эндопротеза тазобедренного сустава выглядит следующим образом: Class III (Rule 8).
В то же время, если мы проведем поиск по базе FDA, то получим следующую картину:
Как мы видим, многие из потенциальных продуктов-аналогов относятся в США к классу II. Если ваш продукт по характеристикам соответствует подобному аналогу, то он также будет классифицирован как класс II. В результате возникнет ситуация, когда в Европе медицинское изделие попадает в класс III, а в США в класс II
Существует отдельная категория медицинских изделий, которые изготавливаются по индивидуальному заказу для каждого отдельного пациента (custom-made devices). В принципе, это означает, что такие изделия не являются результатом серийного производства. А потому на них не распространяются многие требования, предъявляемые к массово производимым изделиям.
Такие индивидуальные изделия не требуют получения одобрения регуляторных органов перед использованием в клинических условиях ни в ЕС, ни в США. Впрочем, это не означает, что медицинские изделия индивидуального назначения могут применяться без всякого контроля. И в Европе, и в США имеются особые регуляторные процедуры, распространяющиеся на эту категорию продуктов. И в обеих юрисдикциях индивидуальные изделия должны сопровождаться техническими файлами.
Документ, отражающий класс продукта и способ определения этого класса, является частью любого технического файла. Поэтому даже для медицинских изделий для индивидуального применения установление класса - необходимое условие. Ведь класс изделий определяет, среди прочего, степень внимания и осторожности, с которыми медицинский и технический персонал должен обращаться с ними.
И в этом есть смысл. Так, мы ранее говорили об эндопротезе тазобедренного сустава. Тот факт, что он в ЕС, а в ряде случаев и в США, относится к высшему классу риска должен заставить медицинский и технический персонал проявлять высшую степень осторожности при работе с ним.
Эндопротез может быть изготовлен и индивидуально для конкретного пациента. И тот факт, что продукт не является результатом серийного производства, нисколько не снижает риски, связанные с его применением. Определение класса позволяет взять эти риски под контроль и обращаться с изделием соответствующим образом.
Скачать эту статью в PDF-формате
1. European Union, 2017. Medical Device Regulation (Regulation (EU) 2017/745)
2. US Code of Federal Regulations Title 21 Part 860.



Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.

+357 22253765
info@mdrc-consulting.com

Европейский Союз и США - два крупнейших рынка медицинских изделий. Ни один производитель не откажется вывести свою продукцию на один из них - а еще лучше на оба. Но, как известно, эти рынки строго регулируются. А значит, чтобы на них попасть, необходимо пройти определенные регуляторные процедуры.
Логично предположить, что разные медицинские изделия требуют несколько разных подходов. Было бы странно, если бы, скажем, к эластичному бинту и к вентрикулоперитонеальному шунту предъявлялись одинаковые требования. И здесь нам на помощь приходит классификация медицинских изделий, которая и определяет регуляторные пути, которыми придется пройти вашему продукту, прежде чем он окажется на рынке и принесет вашей компании миллионы.
Для того чтобы понять, какова будет процедура получения одобрения вашего продукта (а значит каковы будут сроки одобрения и каких затрат потребует этот процесс), необходимо определить, к какому классу риска он относится. О том, как это сделать, и пойдет речь в данной брошюре.
Скачать эту статью в PDF-формате
Здесь следует заметить, что американский и европейский подходы весьма существенно различаются. Мы разберем оба этих подхода и рассмотрим две классификации на конкретных примерах медицинских изделий. Но сначала давайте разберемся с важнейшим вопросом, над которым, почему-то, производители медицинских изделий стараются не задумываться. А именно: на каком этапе следует обращаться к теме классификации?
Многие компании, разрабатывающие медицинскую продукцию, откладывают определение класса продукта на конец разработки. Мол, не будем городить огород - сначала создадим изделие, а там уж определим, каким именно образом мы будем выводить его на рынок. Ответственно заявляем: это плохой подход!
Вам следует определить класс изделия как можно раньше - желательно в самом начале разработки, на этапе создания идеи, а затем контролировать возможные изменения класса продукта. Дело в том, что, если вы этого не сделаете, то в итоге вы можете получить изделие более высокого класса риска, чем вам представлялось вначале. А временные и финансовые затраты на регистрацию продукта по мере увеличения класса риска возрастают в геометрической прогрессии.
В результате вместо трех-четырех месяцев и нескольких тысяч долларов получение одобрения может потребовать пару лет и затрат, измеряемых сотнями тысяч. Вполне возможно, что компания окажется не готова к таким радикальным изменениям плана и бюджета разработки. И в итоге продукт, который мог бы продаваться в Европе и США и приносить компании колоссальные прибыли, навсегда остается на локальном рынке. А то и вовсе разработка будет остановлена.
Мораль проста: классификация медицинского изделия - это такой же управляемый элемент проекта разработки, как таймлайны, бюджет, качество или человеческие ресурсы. Но для того, чтобы управлять классификацией, вам необходимо определить класс будущего изделия как можно раньше и соответствующим образом планировать ресурсы.
Все это не значит, что в процессе разработки класс не может меняться. Вполне возможно, что класс финального продукта будет отличаться от класса, определенного исходно. Но это будет осознанное решение, принятое с учетом оценки проектных рисков и необходимости пересмотра ресурсов.
Как мы уже упомянули, регуляторные органы ЕС и США по-разному подходят к вопросам классификации медицинских изделий. В ЕС выделяю четыре класса медицинских изделий - I, IIa, IIb, III. В США - три: I, II, III.
Европейская классификация основана на правилах. Правила классификации приводятся в нормативном документе ЕС, известном как Medical Device Regulation (MDR), он же Регламент ЕС 2017/745 (Regulation (EU) 2017/745). Данный документ находится в свободном доступе.
Пояснение: В данной статье мы не рассматриваем европейское законодательство, касающееся изделий для in vitro диагностики. Правила классификации для продукции in vitro изложены в нормативном документе In Vitro Medical Device Regulation (IVDR) (Regualtion 2017/746).
Всего таких правил - 22. Чтобы определить класс вашего продукта, вам необходимо проверить его соответствие каждому из правил, начиная с Правила 1. Когда вы пройдете все применимые к вашему продукту правила, вам должно стать понятно, к какому классу относится ваш продукт.
Правила разбиты на 4 группы. Первая группа относится к неинвазивным изделиям. Вторая - к инвазивным. Третья - к активным (т.е. приводимым в движение каким-либо источником энергии). Четвертая - специальные правила, применимые к отдельным категориям продукции.
В отличие от европейских регуляторов (европейская система децентрализована, поэтому говорить о едином европейском регуляторе было бы некорректно), FDA определяет класс каждого изделия и хранит его индивидуальные данные в гигантской базе данных (Принципы классификации изложены в нормативе 21 CFR Part 860). Чтобы определить класс вашего изделия, вам необходимо обратиться к этой базе данных (доступ к ней имеет любой пользователь интернета) и найти продукт, аналогичный вашему. Класс вашего продукта будет соответствовать классу аналога. Звучит просто. Однако на практике все выглядит несколько сложнее.
Проблема в том, что база данных FDA действительно гигантская, и, чтобы найти в ней аналог вашего изделия, нужно использовать правильные поисковые слова. Поэтому определение класса медицинского изделия в США начинается с составления списка поисковых слов. При этом вам необходимо учитывать правильность написания и особенности американской терминологии, которая может отличаться от терминологии, к которой привыкли европейцы.
Чтобы получить представление о том, как определяется класс медицинского изделия в Европейском Союзе, обратимся к примерам двух продуктов. Пусть оба продукта относятся к области ортопедии: хирургическое сверло и ортопедическая стелька. Проверим оба продукта на соответствие правилам европейской классификации, изложенным в Приложении VIII Регламента ЕС 2017/745 (чтобы лучше понять то, о чем идет речь ниже, хорошо иметь текст приложения перед глазами).
Первые четыре правила относятся только к неинвазивным изделиям. Хирургическое сверло - инвазивное изделие. Поэтому для определения класса следует сразу перейти к разделу "Инвазивные изделия", который начинается с Правила 5.
Правило 5 относится к инвазивным изделиям, проникающим в тело человека через естественные отверстия. Поэтому к сверлу оно также не применимо, поскольку добраться до кости, не нарушив целостности тканей невозможно. В свою очередь, Правило 6 относится к хирургическим инвазивным (нарушающим целостность тканей) изделиям, значит, сверло подпадает под действие этого правила.
Правило 6 гласит: все хирургические инвазивные изделия, применяемые краткосрочно (менее 60 минут) относятся к классу IIa. Соответственно, сверло должно было бы относиться к классу IIa. Однако у Правила 6 имеются оговорки, которые приводятся в виде списка под текстом правила. Одна из оговорок гласит, что все многоразовые хирургические инструменты относятся к классу I.
Это хорошая новость для производителя, но надо проверить изделие на соответствие и другим правилам. После проверки мы устанавливаем, что хирургическое сверло не подпадает под действие других правил. Соответственно данный продукт относится к классу I.
Изделия класса I делятся на обычный класс I, а также на стерильные изделия класса I и изделия класса I с измерительными функциями. Регуляторные процедуры для двух последних категорий несколько сложнее и строже.
Сверло не имеет измерительной функции - из этой категории мы можем его сразу исключить. Что касается стерильности - безусловно в условиях операционной сверло используется в стерильном состоянии. Но для классификации важно не это, а то, в каком состоянии оно поставляется покупателям.
Многоразовое сверло поставляется в нестерильном состоянии. Поэтому в категорию стерильных изделий класса I оно не попадает. В итоге получаем изделие, относящееся к самому низкому классу риска - класс I (нестерильные, без функции измерения).
Теперь определим класс ортопедической стельки. Это неинвазивное изделие. Соответственно, классификацию начинаем с раздела «Неинвазивные изделия», т.е. с Правила 1.
Правило 1 гласит: Все неинвазивные изделия относятся к классу I, если все дальнейшие правила неприменимы к ним. Проверяем все дальнейшие правила и убеждаемся, что к ортопедической стельке они неприменимы. Следовательно, стелька относится к классу I. Поскольку она не используется для измерений и не поставляется в стерильном виде, получаем класс 1 (нестерильные, без функции измерения).
Таким образом, и хирургическое сверло, и ортопедическая стелька относятся к классу I (нестерильные, без функции измерения). Регуляторная процедура для таких изделий будет самая простая. Фактически такой продукт может выводиться на рынок без разрешения регуляторов (нотифицированных организаций).
Тем не менее мы пришли к подобной классификации двух изделий несколько разными путями. Для хирургического сверла определяющим стало Правило 5, для ортопедической стельки - Правило 1. В соответствующем разделе технического файла класс изделия приводится с указанием определяющего правила.
Например:
Device: Drill bit
Class I (Rule 5)
Device: Orthotic shoe insert
Class I (Rule 1).
Теперь рассмотрим классификацию медицинских изделий в США. К какому классу будут относиться, скажем, костные кусачки. Для того чтобы определить класс, нам потребуется найти продукт-аналог (predicate device), который был ранее зарегистрирован FDA и находится в базе данных регулятора.
Для этого загружаем базу данных и осуществляем поиск по запросу "wire cutter". В итоге находим ранее зарегистрированный продукт, который может выступать в роли аналога.
Продукт-аналог относится к классу I. Значит и ваш продукт относится к классу I. В базе данных FDA содержится и другая полезная информация о продукте-аналоге. Пройдя по ссылкам, приведенным в результатах поиска можно получить подробное описание продукта или его дженериковой группы, а также данные о проблемах с продуктом, отзывах и т.д. Изучив информацию, касающуюся продукта-аналога, можно установить является ли ваш продукт исключением из 510(k), а также должен ли он непременно производиться в условиях GMP или, опять же, является исключением.
Итак, полная классификация костных кусачек в США будет выглядеть следующим образом:
Device: Cutter, Wire
Class I. Product Code: HTW. Regulation No.: 888.4540
Аналогичным образом осуществляется классификация любого медицинского изделия. Например, уже знакомого нам хирургического сверла.
Как правило, поиск выдает несколько продуктов, которые могли бы выступить в роли аналога для вашего изделия. Выбор продукта-аналога осуществляется самим производителем, который отвечает за правильность этого выбора. При этом производитель должен руководствоваться характеристиками своего изделия и продуктов-аналогов.
Мы рассмотрели примеры изделий, относящихся к классу I и в ЕС, и в США. Тем не менее классы изделий в двух данных юрисдикциях совпадают далеко не всегда. Европейские классы IIа и IIb совсем необязательно соответствуют американскому классу II. Точно так же могут не совпадать и классы I и III. Рассмотрим пример эндопротеза тазобедренного сустава.
Правило 8 европейской классификации гласит: Все имплантируемые изделия, а также хирургические инвазивные изделия для долгосрочного применения относятся к классу IIb...
Далее приводится ряд оговорок, среди которых есть и такая: если только они не являются частичными или полными заменителями суставов. В последнем случае они относятся к классу III (за исключением винтов, клиньев, пластин). Соответственно, классификация эндопротеза тазобедренного сустава выглядит следующим образом: Class III (Rule 8).
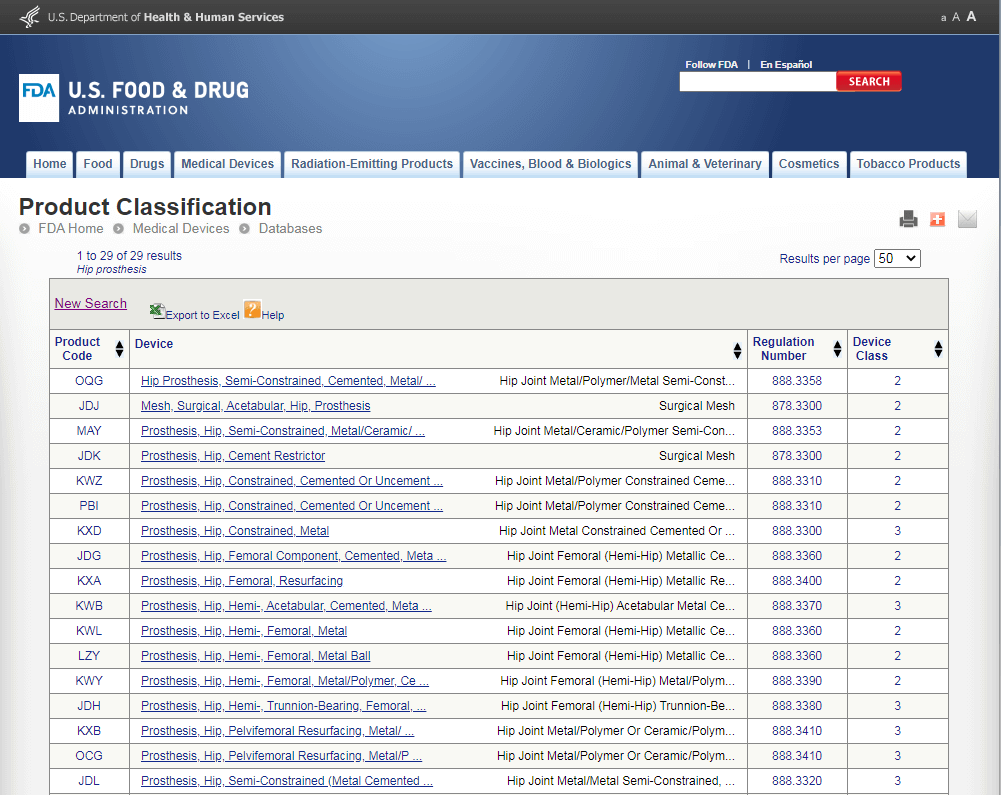
В то же время, если мы проведем поиск по базе FDA, то получим следующую картину:
Как мы видим, многие из потенциальных продуктов-аналогов относятся в США к классу II. Если ваш продукт по характеристикам соответствует подобному аналогу, то он также будет классифицирован как класс II. В результате возникнет ситуация, когда в Европе медицинское изделие попадает в класс III, а в США в класс II
Существует отдельная категория медицинских изделий, которые изготавливаются по индивидуальному заказу для каждого отдельного пациента (custom-made devices). В принципе, это означает, что такие изделия не являются результатом серийного производства. А потому на них не распространяются многие требования, предъявляемые к массово производимым изделиям.
Такие индивидуальные изделия не требуют получения одобрения регуляторных органов перед использованием в клинических условиях ни в ЕС, ни в США. Впрочем, это не означает, что медицинские изделия индивидуального назначения могут применяться без всякого контроля. И в Европе, и в США имеются особые регуляторные процедуры, распространяющиеся на эту категорию продуктов. И в обеих юрисдикциях индивидуальные изделия должны сопровождаться техническими файлами.
Документ, отражающий класс продукта и способ определения этого класса, является частью любого технического файла. Поэтому даже для медицинских изделий для индивидуального применения установление класса - необходимое условие. Ведь класс изделий определяет, среди прочего, степень внимания и осторожности, с которыми медицинский и технический персонал должен обращаться с ними.
И в этом есть смысл. Так, мы ранее говорили об эндопротезе тазобедренного сустава. Тот факт, что он в ЕС, а в ряде случаев и в США, относится к высшему классу риска должен заставить медицинский и технический персонал проявлять высшую степень осторожности при работе с ним.
Эндопротез может быть изготовлен и индивидуально для конкретного пациента. И тот факт, что продукт не является результатом серийного производства, нисколько не снижает риски, связанные с его применением. Определение класса позволяет взять эти риски под контроль и обращаться с изделием соответствующим образом.
Скачать эту статью в PDF-формате
1. European Union, 2017. Medical Device Regulation (Regulation (EU) 2017/745)
2. US Code of Federal Regulations Title 21 Part 860.
Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.
+357 22253765
info@mdrc-consulting.com
Мы будем рады обсудить ваш новый проект!