510(k) - это уведомительная регуляторная процедура, направленная на получение одобрения FDA (если соблюдать терминологическую точность, на установление отсутствия возражений со стороны регулятора) на выведение изделия медицинского назначения на рынок США. Наряду с продуктами-исключениями (510(k) exempts) и разрешительной процедурой Premarket Approval (PMA), 510(k) представляет собой один из трех регуляторных путей вывода медицинской продукции на американский рынок. Грубо говоря, 510(k) - это один из вариантов регистрации изделий медицинского назначения в США. Причем основная часть медицинских продуктов на американском рынке подпадают именно под требования 510(k), а не двух других процедур. Большая часть продукции класса II, а также ряд продуктов классов I и III должны пройти именно 510(k). В данной статье рассматривается, что такое 510(k), какие существуют виды этой процедуры, и какие условия производитель должен выполнить, чтобы успешно ее пройти.
Существенная эквивалентность (Substantial Equivalence) - это центральная концепция американской регуляторной системы и метод, используемый FDA для доказательства того, что новый продукт, выводимый на рынок, по своим безопасности и эффективности является сопоставимым с продуктом, уже находящимся на рынке США (Predicate Device - продукт-предшественник или продукт-аналог).
В целом, здесь работает простое правило. Если продукт-предшественник из базы данных FDA при его размещении на рынке требовал регуляторной процедуры 510(k), то и новый продукт требует этой процедуры.
Это не значит, что ваш продукт и продукт-предшественник должны быть идентичными. Возможны отличия. Существенная эквивалентность устанавливается на основе анализа целого ряда факторов: устройство изделия, материалы, производственные процессы, технические и функциональные характеристики, безопасность, эффективность, маркировка и др.
Чтобы понять, что такое 510(k) с практической точки зрения, необходимо рассмотреть три варианта этой регуляторной процедуры. Каждая из них требует от производителя различного времени и различных ресурсов. Можно выделить следующие варианты [1]:
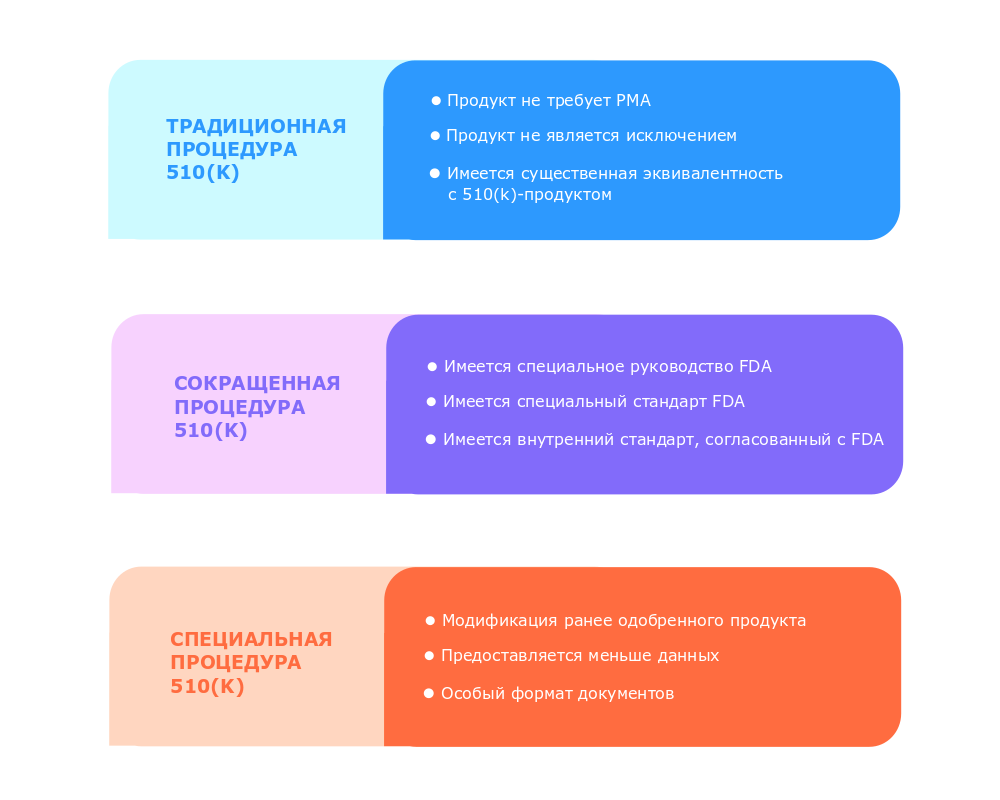
Эту процедуру проходит большинство компаний, выводящих новые продукты, относящиеся к классу II, на рынок США. Она требуется тогда, когда:
Традиционная процедура длится 90 дней.[1]
Специальная процедура применяется, когда производитель вносит какие-либо изменения в устройство своего продукта, ранее получившего одобрение FDA, или когда создается модификация продукта. При этом возможны только ограниченные изменения, касающиеся целевого назначения продукта и технологий, использующихся в нем. Во время прохождения специальной процедуры FDA требует меньше данных относительно функциональных характеристик и меньший объем доказательств существенной эквивалентности продукта.
При недостаточности доказательств существенной эквивалентности новой модификации продукта, FDA может потребовать проведения традиционной процедуры.
Продолжительность специальной процедуры составляет 30 дней.[1]
Сокращенная процедура применяется тогда, когда имеются специальные руководства и стандарты FDA, касающиеся данного типа продукции. Процедура требует меньший объем документации. Производитель может выбрать сокращенную процедуру, если:
Сокращенная процедура 510(k) длится 90 дней.[1]
Существует целый ряд вариантов и сценариев при подаче 510(k). Для FDA каждая регуляторная подача уникальна. Поэтому точно предсказать, как будут развиваться события после того, как вы отправите документацию американскому регулятору, практически невозможно. Для некоторых компаний и продуктов нахождение и доказательство Существенной Эквивалентности может стать вызовом. Непродуманные действия в такой ситуации могут привести к задержкам, дополнительным тратам и даже стать причиной отказа.
Создание регуляторной стратегии поможет вам сделать ваши подачи более организованными и предсказуемыми. Грамотно выстроенная стратегия станет ключом к беспрепятственному прохождению всех регуляторных барьеров, стоящих между производителем и весьма привлекательным американским рынком изделий медицинского назначения.
1. 510(k) Submission Programs. U.S. Food and Drug Administration.



Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.

+357 22253765
info@mdrc-consulting.com
510(k) - это уведомительная регуляторная процедура, направленная на получение одобрения FDA (если соблюдать терминологическую точность, на установление отсутствия возражений со стороны регулятора) на выведение изделия медицинского назначения на рынок США. Наряду с продуктами-исключениями (510(k) exempts) и разрешительной процедурой Premarket Approval (PMA), 510(k) представляет собой один из трех регуляторных путей вывода медицинской продукции на американский рынок. Грубо говоря, 510(k) - это один из вариантов регистрации изделий медицинского назначения в США. Причем основная часть медицинских продуктов на американском рынке подпадают именно под требования 510(k), а не двух других процедур. Большая часть продукции класса II, а также ряд продуктов классов I и III должны пройти именно 510(k). В данной статье рассматривается, что такое 510(k), какие существуют виды этой процедуры, и какие условия производитель должен выполнить, чтобы успешно ее пройти.
Существенная эквивалентность (Substantial Equivalence) - это центральная концепция американской регуляторной системы и метод, используемый FDA для доказательства того, что новый продукт, выводимый на рынок, по своим безопасности и эффективности является сопоставимым с продуктом, уже находящимся на рынке США (Predicate Device - продукт-предшественник или продукт-аналог).
В целом, здесь работает простое правило. Если продукт-предшественник из базы данных FDA при его размещении на рынке требовал регуляторной процедуры 510(k), то и новый продукт требует этой процедуры.
Это не значит, что ваш продукт и продукт-предшественник должны быть идентичными. Возможны отличия. Существенная эквивалентность устанавливается на основе анализа целого ряда факторов: устройство изделия, материалы, производственные процессы, технические и функциональные характеристики, безопасность, эффективность, маркировка и др.
Чтобы понять, что такое 510(k) с практической точки зрения, необходимо рассмотреть три варианта этой регуляторной процедуры. Каждая из них требует от производителя различного времени и различных ресурсов. Можно выделить следующие варианты[1]:
Эту процедуру проходит большинство компаний, выводящих новые продукты, относящиеся к классу II, на рынок США. Она требуется тогда, когда:
Традиционная процедура длится 90 дней.[1]
Специальная процедура применяется, когда производитель вносит какие-либо изменения в устройство своего продукта, ранее получившего одобрение FDA, или когда создается модификация продукта. При этом возможны только ограниченные изменения, касающиеся целевого назначения продукта и технологий, использующихся в нем. Во время прохождения специальной процедуры FDA требует меньше данных относительно функциональных характеристик и меньший объем доказательств существенной эквивалентности продукта.
При недостаточности доказательств существенной эквивалентности новой модификации продукта, FDA может потребовать проведения традиционной процедуры.
Продолжительность специальной процедуры составляет 30 дней.[1]
Сокращенная процедура применяется тогда, когда имеются специальные руководства и стандарты FDA, касающиеся данного типа продукции. Процедура требует меньший объем документации. Производитель может выбрать сокращенную процедуру, если:
Сокращенная процедура 510(k) длится 90 дней.[1]
Существует целый ряд вариантов и сценариев при подаче 510(k). Для FDA каждая регуляторная подача уникальна. Поэтому точно предсказать, как будут развиваться события после того, как вы отправите документацию американскому регулятору, практически невозможно. Для некоторых компаний и продуктов нахождение и доказательство Существенной Эквивалентности может стать вызовом. Непродуманные действия в такой ситуации могут привести к задержкам, дополнительным тратам и даже стать причиной отказа.
Создание регуляторной стратегии поможет вам сделать ваши подачи более организованными и предсказуемыми. Грамотно выстроенная стратегия станет ключом к беспрепятственному прохождению всех регуляторных барьеров, стоящих между производителем и весьма привлекательным американским рынком изделий медицинского назначения.
1. 510(k) Submission Programs. U.S. Food and Drug Administration.
Мы здесь, чтобы помочь вам вывести вашу продукцию медицинского назначения на внешние рынки.
+357 22253765
info@mdrc-consulting.com
Мы будем рады обсудить ваш новый проект!