MDRC assists in vitro medical device companies with registration, representation, quality system compliance and distribution in Europe as shown below.
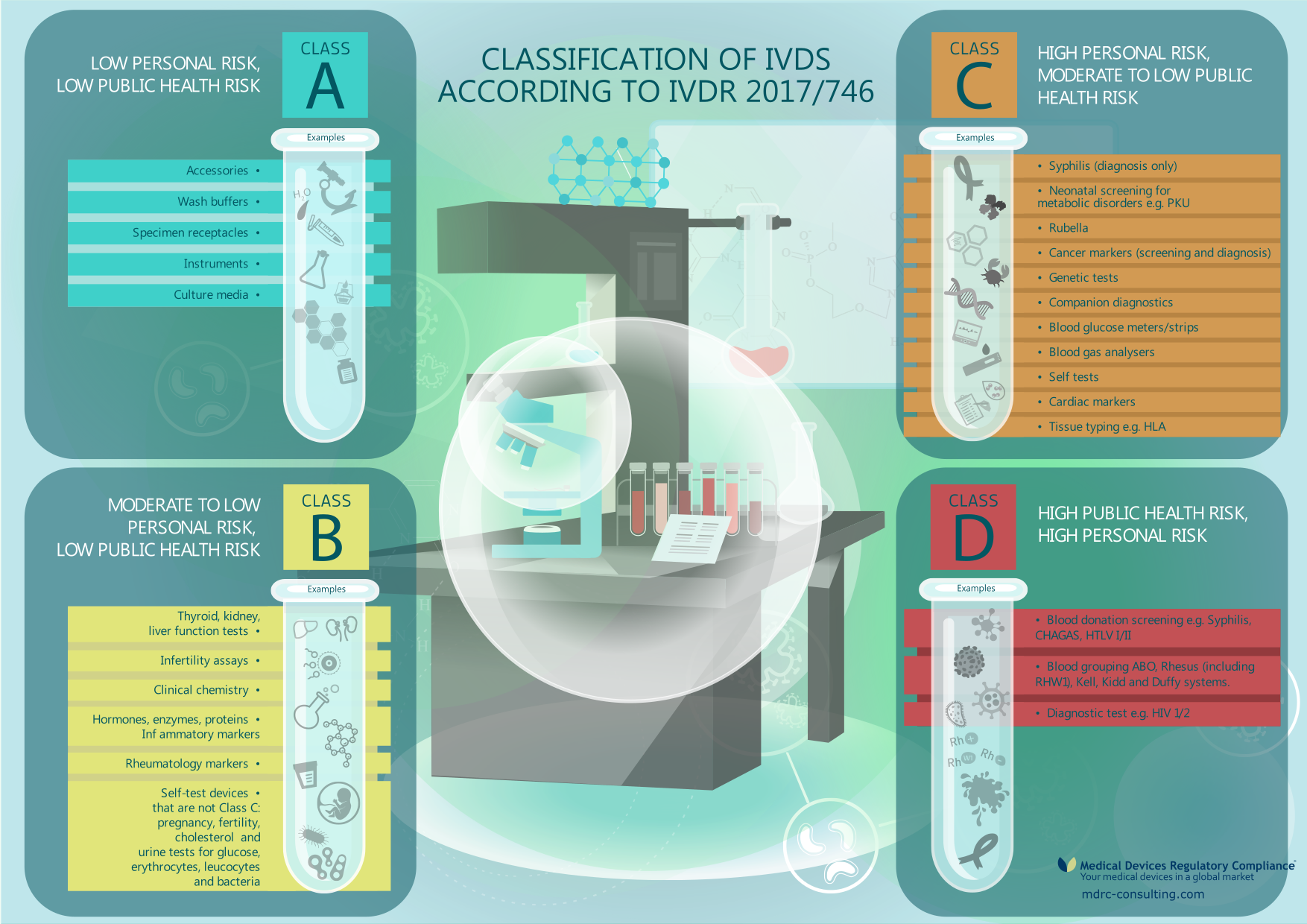
In Europe, in vitro medical devices are classified using a series of rules found in Annex VIII of the In Vitro Diagnostic Medical Devices Regulation (IVDR). Therefore, a product considered certain class in China, Russia, USA or Canada might carry a different classification in Europe. Proper evaluation of classification criteria is important and we can assist you in this process.
Technical Files are required for all device classes. Design Dossiers are needed for Class III devices and are much like a US FDA PMA. We have compiled Technical Files and Design Dossiers for a wide range of in vitro medical devices so are fully prepared to help you obtain your CE Marking certificate. We can also assist with ISO 14971, clinical data evaluations, labeling reviews and more.
MDRC has procedures in place for implementing quality management systems that address regulatory requirements in most parts of the world. Most manufacturers implement a Quality Management System by applying the ISO 13485 standard and we can assist you with implementation. If you already have a US FDA QSR compliant quality system, we can modify your existing system to meet European QMS requirements and ensure compliance with Brazilian, Mexican and other requirements as desired.
Companies without a location in Europe must appoint an Authorized Representative to act on their behalf. Although a local distributor can fulfill this role, hiring a professional, independent EC REP gives you the freedom to switch distributors at any time. This is particularly important given that the EC REP name must appear on your labeling throughout Europe. We can help you find a Representative or act as an independent Authorized Representative for you.

Both Medical Device Regulation – MDR (EU 2017/745) and In Vitro Diagnostic Regulation – IVDR (EU 2017/746) require companies to have a Person Responsible for Regulatory Compliance (PRRC) at their disposal.
Manufacturers of high risk in vitro diagnostic devices are audited each year by Notified Bodies. We can conduct pre-assessment or maintenance audits prior to Notified Body audits as needed. We can also provide onsite training on CE Marking, the Medical Devices Directive and internal auditing for your employees.
With 31 countries and 20+ official languages, qualifying distributors in Europe can be a challenge. We can find, analyze, select and manage the right distributors in specific countries and ensure that they are fully committed to selling your devices.
MDRC assists in vitro medical device companies with registration, representation, quality system compliance and distribution in Europe as shown below.
In Europe, in vitro medical devices are classified using a series of rules found in Annex VIII of the In Vitro Diagnostic Medical Devices Regulation (IVDR). Therefore, a product considered certain class in China, Russia, USA or Canada might carry a different classification in Europe. Proper evaluation of classification criteria is important and we can assist you in this process.
Technical Files are required for all device classes. Design Dossiers are needed for Class III devices and are much like a US FDA PMA. We have compiled Technical Files and Design Dossiers for a wide range of in vitro medical devices so are fully prepared to help you obtain your CE Marking certificate. We can also assist with ISO 14971, clinical data evaluations, labeling reviews and more.
MDRC has procedures in place for implementing quality management systems that address regulatory requirements in most parts of the world. Most manufacturers implement a Quality Management System by applying the ISO 13485 standard and we can assist you with implementation. If you already have a US FDA QSR compliant quality system, we can modify your existing system to meet European QMS requirements and ensure compliance with Brazilian, Mexican and other requirements as desired.
Companies without a location in Europe must appoint an Authorized Representative to act on their behalf. Although a local distributor can fulfill this role, hiring a professional, independent EC REP gives you the freedom to switch distributors at any time. This is particularly important given that the EC REP name must appear on your labeling throughout Europe. We can help you find a Representative or act as an independent Authorized Representative for you.
Both Medical Device Regulation – MDR (EU 2017/745) and In Vitro Diagnostic Regulation – IVDR (EU 2017/746) require companies to have a Person Responsible for Regulatory Compliance (PRRC) at their disposal.
Manufacturers of high risk in vitro diagnostic devices are audited each year by Notified Bodies. We can conduct pre-assessment or maintenance audits prior to Notified Body audits as needed. We can also provide onsite training on CE Marking, the Medical Devices Directive and internal auditing for your employees.
With 31 countries and 20+ official languages, qualifying distributors in Europe can be a challenge. We can find, analyze, select and manage the right distributors in specific countries and ensure that they are fully committed to selling your devices.