
Medical device companies seeking to enter the market of Costa Rica must demonstrate regulatory compliance with local regulations and register their products with the national regulatory authorities.
Registration and control of medical devices in Costa Rica is the direct responsibility of the Ministry of Health of Costa Rica - Ministerio de Salud de Costa Rica.
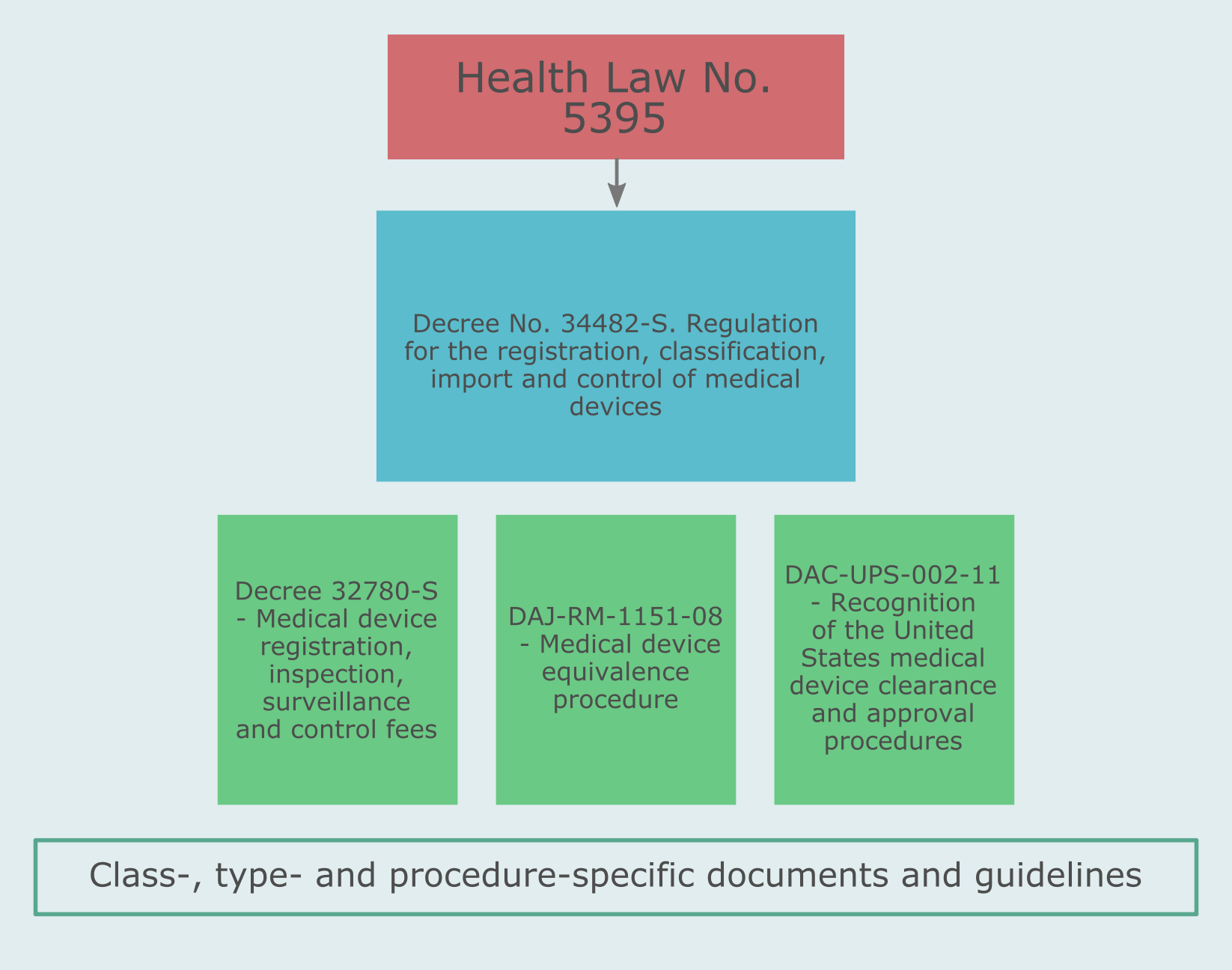
There are a number of regulations applicable to medical devices and relevant for the registration of medical devices in Costa Rica:
In Costa Rica medical devices fall under the definition of "Biomedical equipment and material" (Equipo y material biomédico - EMB).
EMB is defined as instrument, device, equipment, material, software or another item, used alone or in combination and intended by the manufacturer to be used in humans with any of the following purposes: diagnosis, prevention, control, treatment, alleviation, relief or compensation of a disease, injury or deficiency; investigation, replacement or modification of anatomical structures or of physiological processes; conception control - provided that the product does not exert its function by pharmacological, immunological or metabolic mechanisms.
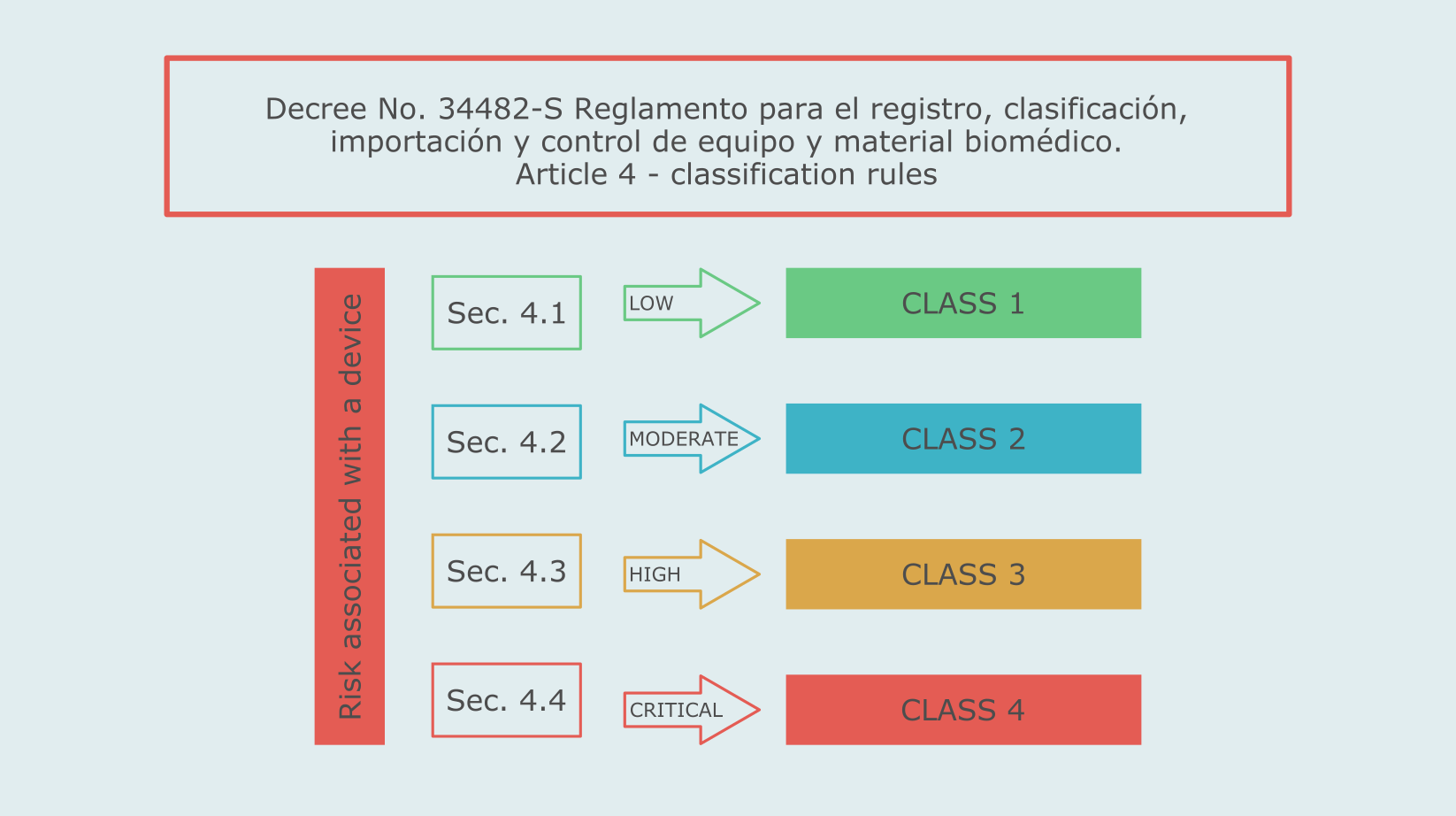
Costa Rica has a medical device classification system similar to Health Canada’s classification. The classification rules are set forth in article 4 of the Regulation for the registration, classification, import and control of medical devices (Decree No. 34482-S).
Class 1 medical devices in Costa Rica are exempt from the registration procedure. Class 2 products have a simplified registration procedure, while Class 3 and 4 devices will have higher requirements, such as clinical studies.
Class 3 and 4 devices that are listed, cleared, or approved with the US FDA are eligible for the simplified registration procedure, which is similar to the Class 2 requirements.
The documents required for the medical device registration are listed in the regulation Para el registro de Equipo y Material Biomédico (EMB) issued by the Ministry of Health. The registration dossiers differ from class to class.
In addition to the documents listed above:
In addition to the documents submitted for all classes:
In addition to the documents submitted for all classes:
The Ministry of Health reviews the registration documentation in two phases:
The applicant submits to the Ministry of Health the registration request with some of the registration documentation listed above.
The adequacy and legal compliance of documentation is assessed within a period of fifteen days class 2 devices and a period of thirty days for class 3 and 4 devices.
If the review outcome is positive, the applicant receives the Approval of Phase 1.
The applicant submits the full registration dossier and the Approval of Phase 1. If the technical review demonstrates compliance with the requirements of the Ministry of Health, the applicant receives a Registration Certificate, which is valid for 5 years.

We are here to help you place your medical devices on your strategic markets.

+49 176 67510274
info@mdrc-consulting.com
Medical device companies seeking to enter the market of Costa Rica must demonstrate regulatory compliance with local regulations and register their products with the national regulatory authorities.
Registration and control of medical devices in Costa Rica is the direct responsibility of the Ministry of Health of Costa Rica - Ministerio de Salud de Costa Rica.
There are a number of regulations applicable to medical devices and relevant for the registration of medical devices in Costa Rica:
In Costa Rica medical devices fall under the definition of "Biomedical equipment and material" (Equipo y material biomédico - EMB).
EMB is defined as instrument, device, equipment, material, software or another item, used alone or in combination and intended by the manufacturer to be used in humans with any of the following purposes: diagnosis, prevention, control, treatment, alleviation, relief or compensation of a disease, injury or deficiency; investigation, replacement or modification of anatomical structures or of physiological processes; conception control - provided that the product does not exert its function by pharmacological, immunological or metabolic mechanisms.
Costa Rica has a medical device classification system similar to Health Canada’s classification. The classification rules are set forth in article 4 of the Regulation for the registration, classification, import and control of medical devices (Decree No. 34482-S).
Class 1 medical devices in Costa Rica are exempt from the registration procedure. Class 2 products have a simplified registration procedure, while Class 3 and 4 devices will have higher requirements, such as clinical studies.
Class 3 and 4 devices that are listed, cleared, or approved with the US FDA are eligible for the simplified registration procedure, which is similar to the Class 2 requirements.
The documents required for the medical device registration are listed in the regulation Para el registro de Equipo y Material Biomédico (EMB) issued by the Ministry of Health. The registration dossiers differ from class to class.
In addition to the documents listed above:
In addition to the documents submitted for all classes:
In addition to the documents submitted for all classes:
The Ministry of Health reviews the registration documentation in two phases:
The applicant submits to the Ministry of Health the registration request with some of the registration documentation listed above.
The adequacy and legal compliance of documentation is assessed within a period of fifteen days class 2 devices and a period of thirty days for class 3 and 4 devices.
If the review outcome is positive, the applicant receives the Approval of Phase 1.
The applicant submits the full registration dossier and the Approval of Phase 1. If the technical review demonstrates compliance with the requirements of the Ministry of Health, the applicant receives a Registration Certificate, which is valid for 5 years.
We are here to help you place your medical devices on your strategic markets.
+49 176 67510274
info@mdrc-consulting.com

